 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3hha | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of cathepsin L in complex with AZ12878478 | ||||||
 Components Components | Cathepsin L1 | ||||||
 Keywords Keywords | HYDROLASE / PROTEROS BIOSTRUCTURES GMBH / Cathepsin L / Inhibitors / Disulfide bond / Glycoprotein / Lysosome / Protease / Thiol protease / Zymogen | ||||||
| Function / homology |  Function and homology information Function and homology informationenkephalin processing / cathepsin L / CD4-positive, alpha-beta T cell lineage commitment / macrophage apoptotic process / chromaffin granule / elastin catabolic process / antigen processing and presentation of peptide antigen / RUNX1 regulates transcription of genes involved in differentiation of keratinocytes / endolysosome lumen / cellular response to thyroid hormone stimulus ...enkephalin processing / cathepsin L / CD4-positive, alpha-beta T cell lineage commitment / macrophage apoptotic process / chromaffin granule / elastin catabolic process / antigen processing and presentation of peptide antigen / RUNX1 regulates transcription of genes involved in differentiation of keratinocytes / endolysosome lumen / cellular response to thyroid hormone stimulus / zymogen activation / Trafficking and processing of endosomal TLR / proteoglycan binding / Assembly of collagen fibrils and other multimeric structures / cysteine-type endopeptidase activator activity involved in apoptotic process / protein autoprocessing / Collagen degradation / fibronectin binding / antigen processing and presentation / collagen catabolic process / serpin family protein binding / cysteine-type peptidase activity / Attachment and Entry / positive regulation of apoptotic signaling pathway / endocytic vesicle lumen / collagen binding / MHC class II antigen presentation / Degradation of the extracellular matrix / multivesicular body / proteolysis involved in protein catabolic process / lysosomal lumen / Endosomal/Vacuolar pathway / antigen processing and presentation of exogenous peptide antigen via MHC class II / histone binding / collagen-containing extracellular matrix / receptor-mediated endocytosis of virus by host cell / Attachment and Entry / adaptive immune response / lysosome / immune response / symbiont entry into host cell / apical plasma membrane / fusion of virus membrane with host plasma membrane / cysteine-type endopeptidase activity / intracellular membrane-bounded organelle / fusion of virus membrane with host endosome membrane / Golgi apparatus / proteolysis / extracellular space / extracellular exosome / extracellular region / nucleus / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.27 Å | ||||||
 Authors Authors | Asaad, N. / Bethel, P.A. / Coulson, M.D. / Dawson, J. / Ford, S.J. / Gerhardt, S. / Grist, M. / Hamlin, G.A. / James, M.J. / Jones, E.V. ...Asaad, N. / Bethel, P.A. / Coulson, M.D. / Dawson, J. / Ford, S.J. / Gerhardt, S. / Grist, M. / Hamlin, G.A. / James, M.J. / Jones, E.V. / Karoutchi, G.I. / Kenny, P.W. / Morley, A.D. / Oldham, K. / Rankine, N. / Ryan, D. / Wells, S.L. / Wood, L. / Augustin, M. / Krapp, S. / Simader, H. / Steinbacher, S. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2009 Title: Dipeptidyl nitrile inhibitors of Cathepsin L. Authors: Asaad, N. / Bethel, P.A. / Coulson, M.D. / Dawson, J.E. / Ford, S.J. / Gerhardt, S. / Grist, M. / Hamlin, G.A. / James, M.J. / Jones, E.V. / Karoutchi, G.I. / Kenny, P.W. / Morley, A.D. / ...Authors: Asaad, N. / Bethel, P.A. / Coulson, M.D. / Dawson, J.E. / Ford, S.J. / Gerhardt, S. / Grist, M. / Hamlin, G.A. / James, M.J. / Jones, E.V. / Karoutchi, G.I. / Kenny, P.W. / Morley, A.D. / Oldham, K. / Rankine, N. / Ryan, D. / Wells, S.L. / Wood, L. / Augustin, M. / Krapp, S. / Simader, H. / Steinbacher, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3hha.cif.gz | 389.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3hha.ent.gz | 332.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3hha.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hh/3hhaftp://data.pdbj.org/pub/pdb/validation_reports/hh/3hha | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | / Major excreted protein / MEP / Cathepsin L1 heavy chain / Cathepsin L1 light chain Mass: 24161.676 Da / Num. of mol.: 4 / Fragment: Heavy chain and light chain: UNP residues 76-333 / Mutation: T223A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSL1, CTSL / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P07711, cathepsin L Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P07711, cathepsin L |
|---|
-Non-polymers , 6 types, 1001 molecules 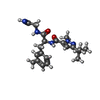





| #2: Chemical | ChemComp-NOW /  Mass: 383.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H29N5O2 Mass: 383.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H29N5O2#3: Chemical | ChemComp-PG4 / | Polyethylene glycol Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#4: Chemical | ChemComp-ACT / | Acetate Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2#5: Chemical | ChemComp-PGE / | Polyethylene glycol Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4#6: Chemical | ChemComp-GOL / | Glycerol Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 993 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | THE MUTATION LISTED IN SEQADV RECORDS HAS BEEN INTRODUCED |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.16 Å3/Da / Density % sol: 43.02 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 7.4 / Details: PEG, pH 7.4, VAPOR DIFFUSION, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Oct 28, 2007 / Details: LN2 cooled fixed-exit |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.27→44.28 Å / Num. all: 182842 / Num. obs: 182842 / % possible obs: 85.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 1.5 % / Rmerge(I) obs: 0.034 |
| Reflection shell | Resolution: 1.27→1.33 Å / Redundancy: 0.4 % / Rmerge(I) obs: 0.098 / % possible all: 65.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 1.27→44.28 Å / Cor.coef. Fo:Fc: 0.98 / Cor.coef. Fo:Fc free: 0.971 / SU B: 1.253 / SU ML: 0.025 / Cross valid method: THROUGHOUT / ESU R: 0.043 / ESU R Free: 0.043 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.158 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.27→44.28 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.27→1.303 Å / Total num. of bins used: 20
|
