Entry Database : PDB / ID : 5k3yTitle Crystal structure of AuroraB/INCENP in complex with BI 811283 Aurora kinase B-A Inner centromere protein A Keywords / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Xenopus laevis (African clawed frog)Method / / Resolution : 1.6 Å Authors Bader, G. / Zahn, S.K. / Zoephel, A. Journal : Mol.Cancer Ther. / Year : 2016Title : Pharmacological Profile of BI 847325, an Orally Bioavailable, ATP-Competitive Inhibitor of MEK and Aurora Kinases.Authors: Sini, P. / Gurtler, U. / Zahn, S.K. / Baumann, C. / Rudolph, D. / Baumgartinger, R. / Strauss, E. / Haslinger, C. / Tontsch-Grunt, U. / Waizenegger, I.C. / Solca, F. / Bader, G. / Zoephel, A. ... Authors : Sini, P. / Gurtler, U. / Zahn, S.K. / Baumann, C. / Rudolph, D. / Baumgartinger, R. / Strauss, E. / Haslinger, C. / Tontsch-Grunt, U. / Waizenegger, I.C. / Solca, F. / Bader, G. / Zoephel, A. / Treu, M. / Reiser, U. / Garin-Chesa, P. / Boehmelt, G. / Kraut, N. / Quant, J. / Adolf, G.R. History Deposition May 20, 2016 Deposition site / Processing site Revision 1.0 Aug 17, 2016 Provider / Type Revision 1.1 Oct 19, 2016 Group Revision 2.0 Nov 20, 2024 Group Advisory / Atomic model ... Advisory / Atomic model / Data collection / Database references / Derived calculations / Polymer sequence / Structure summary Category atom_site / atom_site_anisotrop ... atom_site / atom_site_anisotrop / chem_comp_atom / chem_comp_bond / database_2 / entity / entity_name_com / entity_poly / entity_poly_seq / pdbx_entity_nonpoly / pdbx_entry_details / pdbx_modification_feature / pdbx_nonpoly_scheme / pdbx_poly_seq_scheme / pdbx_struct_assembly_gen / pdbx_struct_mod_residue / pdbx_unobs_or_zero_occ_residues / struct_asym / struct_conn / struct_site / struct_site_gen Item _atom_site.B_iso_or_equiv / _atom_site.Cartn_x ... _atom_site.B_iso_or_equiv / _atom_site.Cartn_x / _atom_site.Cartn_y / _atom_site.Cartn_z / _atom_site.auth_asym_id / _atom_site.auth_atom_id / _atom_site.auth_comp_id / _atom_site.auth_seq_id / _atom_site.group_PDB / _atom_site.label_alt_id / _atom_site.label_asym_id / _atom_site.label_atom_id / _atom_site.label_comp_id / _atom_site.label_entity_id / _atom_site.label_seq_id / _atom_site.occupancy / _atom_site.type_symbol / _atom_site_anisotrop.U[1][1] / _atom_site_anisotrop.U[1][2] / _atom_site_anisotrop.U[1][3] / _atom_site_anisotrop.U[2][2] / _atom_site_anisotrop.U[2][3] / _atom_site_anisotrop.U[3][3] / _atom_site_anisotrop.id / _atom_site_anisotrop.pdbx_auth_asym_id / _atom_site_anisotrop.pdbx_auth_atom_id / _atom_site_anisotrop.pdbx_auth_comp_id / _atom_site_anisotrop.pdbx_auth_seq_id / _atom_site_anisotrop.pdbx_label_alt_id / _atom_site_anisotrop.pdbx_label_asym_id / _atom_site_anisotrop.pdbx_label_atom_id / _atom_site_anisotrop.pdbx_label_comp_id / _atom_site_anisotrop.pdbx_label_seq_id / _atom_site_anisotrop.type_symbol / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _entity_name_com.name / _entity_poly.nstd_monomer / _entity_poly.pdbx_seq_one_letter_code / _entity_poly_seq.mon_id / _pdbx_poly_seq_scheme.auth_mon_id / _pdbx_poly_seq_scheme.auth_seq_num / _pdbx_poly_seq_scheme.mon_id / _pdbx_poly_seq_scheme.pdb_mon_id / _pdbx_struct_assembly_gen.asym_id_list / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_site.details / _struct_site.pdbx_auth_seq_id / _struct_site_gen.label_asym_id
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj Assembly
Assembly


 Enterobacteria phage L1 (virus)
Enterobacteria phage L1 (virus)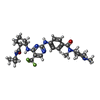
 Mass: 561.642 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C28H38F3N7O2
Mass: 561.642 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C28H38F3N7O2 Mass: 18.015 Da / Num. of mol.: 612 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 612 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X06DA / Wavelength: 0.9801 Å
/ Beamline: X06DA / Wavelength: 0.9801 Å Processing
Processing