 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2j27: The functional role of the conserved active site proline of trios... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2j27 | ||||||
|---|---|---|---|---|---|---|---|
| Title | The functional role of the conserved active site proline of triosephosphate isomerase | ||||||
 Components Components | TRIOSEPHOSPHATE ISOMERASE GLYCOSOMAL | ||||||
 Keywords Keywords |  ISOMERASE / TIM / 2PG / LOOP7 / GLYCOSOME / TIM-BARREL / GLUCONEOGENESIS / LIPID SYNTHESIS / ATOMIC RESOLUTION / GLYCOLYSIS / PENTOSE SHUNT / POINT MUTATION / FATTY ACID BIOSYNTHESIS / 2-PHOSPHO GLYCOLATE / PROTEIN ENGINEERING ISOMERASE / TIM / 2PG / LOOP7 / GLYCOSOME / TIM-BARREL / GLUCONEOGENESIS / LIPID SYNTHESIS / ATOMIC RESOLUTION / GLYCOLYSIS / PENTOSE SHUNT / POINT MUTATION / FATTY ACID BIOSYNTHESIS / 2-PHOSPHO GLYCOLATE / PROTEIN ENGINEERING | ||||||
| Function / homology |  Function and homology informationglycosome / triose-phosphate isomerase / triose-phosphate isomerase activity / gluconeogenesis / glycolytic process / cytoplasm Function and homology informationglycosome / triose-phosphate isomerase / triose-phosphate isomerase activity / gluconeogenesis / glycolytic process / cytoplasmSimilarity search - Function | ||||||
| Biological species |  TRYPANOSOMA BRUCEI BRUCEI (eukaryote) TRYPANOSOMA BRUCEI BRUCEI (eukaryote) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.15 Å | ||||||
 Authors Authors | Casteleijn, M.G. / Alahuhta, M. / Groebel, K. / El-Sayed, I. / Augustyns, K. / Lambeir, A.M. / Neubauer, P. / Wierenga, R.K. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2006 Title: Functional Role of the Conserved Active Site Proline of Triosephosphate Isomerase. Authors: Casteleijn, M.G. / Alahuhta, M. / Groebel, K. / El-Sayed, I. / Augustyns, K. / Lambeir, A.M. / Neubauer, P. / Wierenga, R.K. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" AND "BA" IN EACH CHAIN ON SHEET ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" AND "BA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 9-STRANDED BARREL THIS IS REPRESENTED BY A 10-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2j27.cif.gz | 226.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2j27.ent.gz | 181 KB | Display | PDB format |
| PDBx/mmJSON format | 2j27.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j2/2j27ftp://data.pdbj.org/pub/pdb/validation_reports/j2/2j27 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2j24C  5timS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






































-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.724, -0.69), Vector : |
-Components
| #1: Protein | Mass: 26839.795 Da / Num. of mol.: 2 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) TRYPANOSOMA BRUCEI BRUCEI (eukaryote) / Plasmid: PET3A / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21 / References: UniProt: P04789, triose-phosphate isomerase ESCHERICHIA COLI (E. coli) / Strain (production host): BL21 / References: UniProt: P04789, triose-phosphate isomerase#2: Chemical | 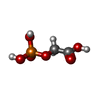  Mass: 156.031 Da / Num. of mol.: 2 Mass: 156.031 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C2H5O6P #3: Chemical | ChemComp-SO4 / | Sulfate  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 686 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 686 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.51 Å3/Da / Density % sol: 51.09 % |
|---|---|
| Crystal grow | pH: 9.5 Details: WELL SOLUTION: 0.1 M CHES PH 9.5, 25 % PEG 1500, 200 MM MGSO4 PROTEIN SOLUTION: 11.5 MG/ML PROTEIN, 20 MM TRIS/HCL PH 7, 100 MM NACL, 1 MM DTT, 1 MM EDTA, 1 MM NAN3 AND 10 MM 2PG |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7A / Wavelength: 0.9023 / Beamline: BW7A / Wavelength: 0.9023 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jun 2, 2005 / Details: RH COATED, ZERODUR |
| Radiation | Monochromator: FIXED EXIT DOUBLE CRYSTAL SI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9023 Å / Relative weight: 1 |
| Reflection | Resolution: 1.15→25 Å / Num. obs: 164758 / % possible obs: 92 % / Observed criterion σ(I): 3 / Redundancy: 6.8 % / Rmerge(I) obs: 0.06 / Net I/σ(I): 25.12 |
| Reflection shell | Resolution: 1.15→1.2 Å / Redundancy: 4.9 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 5.29 / % possible all: 76.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 5TIM Resolution: 1.15→25 Å / Num. parameters: 40486 / Num. restraintsaints: 48408 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 10 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 17876 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.15→25 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
