 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1thl: Thermolysin complexed with a novel glutaramide derivative, n-(1-(... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1thl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Thermolysin complexed with a novel glutaramide derivative, n-(1-(2(r,s)-carboxy-4-phenylbutyl) cyclopentylcarbonyl)-(s)-tryptophan | ||||||
 Components Components | THERMOLYSIN | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / METALLOPROTEINASE / GLUTARAMIDE DERIVATIVE / THERMOLYSIN / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationthermolysin / metalloendopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.7 Å X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.7 Å | ||||||
 Authors Authors | Holland, D.R. / Matthews, B.W. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1994 Title: Inhibition of Thermolysin and Neutral Endopeptidase 24.11 By a Novel Glutaramide Derivative; X-Ray Structure Determination of the Thermolysin-Inhibitor Complex Authors: Holland, D.R. / Barclay, P.L. / Danilewicz, J.C. / Matthews, B.W. / James, K. #1: Journal: Biochemistry / Year: 1984Title: Binding of N-Carboxymethyl Dipeptide Inhibitors to Thermolysin Determined by X-Ray Crystallography: A Novel Class of Transition-State Analogues for Zinc Peptidases Authors: Monzingo, A.F. / Matthews, B.W. #2: Journal: J.Mol.Biol. / Year: 1982Title: Structure of Thermolysin Refined at 1.6 Angstroms Resolution Authors: Holmes, M.A. / Matthews, B.W. #3: Journal: J.Biol.Chem. / Year: 1974Title: The Conformation of Thermolysin Authors: Matthews, B.W. / Weaver, L.H. / Kester, W.R. #4: Journal: Nature New Biol. / Year: 1972Title: Structure of Thermolysin Authors: Matthews, B.W. / Colman, P.M. / Jansonius, J.N. / Titani, K. / Walsh, K.A. / Neura, H. #5: Journal: Nature New Biol. / Year: 1972Title: Three Dimensional Structure of Thermolysin Authors: Matthews, B.W. / Jansonius, J.N. / Colman, P.N. / Schoenborn, B.P. / Duporque, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1thl.cif.gz | 81.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1thl.ent.gz | 59.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1thl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/th/1thlftp://data.pdbj.org/pub/pdb/validation_reports/th/1thl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3tln S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: CIS PROLINE - PRO 51 |
-Components
| #1: Protein | Mass: 34362.305 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-ZN / |   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-0DB / | 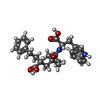  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 476.564 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H32N2O5 Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 476.564 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H32N2O5References: N-({1-[(2S)-2-carboxy-4-phenylbutyl]cyclopentyl}carbonyl)-L-tryptophan #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2OSequence details | THE DENSITY AT POSITION 177 CLEARLY SHOWS RESIDUE GLU, NOT LYS. AUTHORS MAINTAIN THAT THE SWISSPROT ...THE DENSITY AT POSITION 177 CLEARLY SHOWS RESIDUE GLU, NOT LYS. AUTHORS MAINTAIN THAT THE SWISSPROT REFERENCE P00800 IS LIKELY TO BE INCORRECT AT THIS POSITION | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.43 Å3/Da / Density % sol: 49 % |
|---|---|
| Crystal grow | Temperature: 277 K / pH: 7.2 Details: TRIS ACETATE, CALCIUM ACETATE, DMSO, PH 7.2, DILUTION WITH H2O, TEMPERATURE 277K |
| Crystal grow | *PLUS Method: unknown |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: XUONG-HAMLIN MULTIWIRE / Detector: AREA DETECTOR / Date: 1991 / Details: GRAPHITE MONOCHROMATOR, COLLIMATING SLITS |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.58→35 Å / Num. obs: 41090 / % possible obs: 86 % / Observed criterion σ(I): 0 / Biso Wilson estimate: 12.5 Å2 / Rmerge(I) obs: 0.066 |
| Reflection | *PLUS Highest resolution: 1.6 Å / Num. obs: 34103 / Rmerge(I) obs: 0.066 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB ENTRY 3TLN 3tln Resolution: 1.7→10 Å / Isotropic thermal model: ISOTROPIC / σ(F): 0 / Stereochemistry target values: TNT VERSION 1.0
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.7 Å / Lowest resolution: 10 Å / Num. reflection obs: 33922 / Rfactor obs: 0.162 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
