 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1od1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Endothiapepsin PD135,040 complex | ||||||
 Components Components | ENDOTHIAPEPSIN | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / ACID PROTEINASE / INHIBITOR / ASPARTYL PROTEASE / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  CRYPHONECTRIA PARASITICA (chestnut blight fungus) CRYPHONECTRIA PARASITICA (chestnut blight fungus) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.37 Å | ||||||
 Authors Authors | Coates, L. / Erskine, P.T. / Mall, S. / Gill, R.S. / Wood, S.P. / Cooper, J.B. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2003 Title: The Structure of Endothiapepsin Complexed with the Gem-Diol Inhibitor Pd-135,040 at 1.37 A Authors: Coates, L. / Erskine, P.T. / Mall, S. / Williams, P.A. / Gill, R.S. / Wood, S.P. / Cooper, J.B. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1od1.cif.gz | 150 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1od1.ent.gz | 124.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1od1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/od/1od1ftp://data.pdbj.org/pub/pdb/validation_reports/od/1od1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |




















































-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / ASPARTATE PROTEASE / EAPA / EPN-1 Mass: 33813.855 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: ENDOTHIA PARASITICA Source: (natural) CRYPHONECTRIA PARASITICA (chestnut blight fungus)References: UniProt: P11838, endothiapepsin | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-0QS / 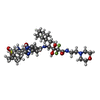  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 784.934 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C36H56F2N7O8S / References: GEM-DIOL INHIBITOR PD-135.040 Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 784.934 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C36H56F2N7O8S / References: GEM-DIOL INHIBITOR PD-135.040 | ||||
| #3: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 418 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 418 / Source method: isolated from a natural source / Formula: H2OCompound details | HYDROLYSES | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.43 Å3/Da / Density % sol: 49.46 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.6 / Details: PH 4.60 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 4.6 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.918056 / Beamline: ID29 / Wavelength: 0.918056 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jan 6, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.918056 Å / Relative weight: 1 |
| Reflection | Resolution: 1.37→10 Å / Num. obs: 68317 / % possible obs: 98.3 % / Redundancy: 6.8 % / Rmerge(I) obs: 0.113 / Net I/σ(I): 3.2 |
| Reflection shell | Resolution: 1.37→1.44 Å / Redundancy: 4 % / Rmerge(I) obs: 0.365 / Mean I/σ(I) obs: 1.7 / % possible all: 95.5 |
| Reflection | *PLUS Highest resolution: 1.37 Å / Lowest resolution: 10 Å / Redundancy: 6.8 % / Rmerge(I) obs: 0.113 |
| Reflection shell | *PLUS % possible obs: 95.5 % / Rmerge(I) obs: 0.365 / Mean I/σ(I) obs: 1.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 1.37→10 Å / Num. parameters: 26565 / Num. restraintsaints: 32311 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 16 / Occupancy sum hydrogen: 2287 / Occupancy sum non hydrogen: 2905 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.37→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.1534 / Rfactor Rwork: 0.1153 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
