 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gkt: Neutron Laue diffraction structure of endothiapepsin complexed wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gkt | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Neutron Laue diffraction structure of endothiapepsin complexed with transition state analogue inhibitor H261 | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / HYDROLASE-HYDROLASE INHIBITOR COMPLEX / PROTEASE-INHIBITOR / ASPARTIC PROTEINASE / HYDROLYSIS / HYDROLASE- HYDROLASE INHIBITOR COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  CRYPHONECTRIA PARASITICA (chestnut blight fungus) CRYPHONECTRIA PARASITICA (chestnut blight fungus)synthetic construct (others) | |||||||||
| Method | NEUTRON DIFFRACTION / NUCLEAR REACTOR / Resolution: 2.1 Å | |||||||||
 Authors Authors | Coates, L. / Erskine, P.T. / Wood, S.P. / Myles, D.A.A. / Cooper, J.B. | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2001 Title: A Neutron Laue Diffraction Study of Endothiapepsin: Implications for the Aspartic Proteinase Mechanism Authors: Coates, L. / Erskine, P.T. / Wood, S.P. / Myles, D.A.A. / Cooper, J.B. | |||||||||
| History |
| |||||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gkt.cif.gz | 136.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gkt.ent.gz | 107.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1gkt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gk/1gktftp://data.pdbj.org/pub/pdb/validation_reports/gk/1gkt | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |














































-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 33795.840 Da / Num. of mol.: 1 / Source method: isolated from a natural source Source: (natural) CRYPHONECTRIA PARASITICA (chestnut blight fungus)References: UniProt: P11838, endothiapepsin |
|---|---|
| #2: Protein/peptide | 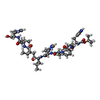  Type: Peptide-like / Class: Inhibitor / Mass: 1103.334 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) / References: H-261 Oligopeptide Type: Peptide-like / Class: Inhibitor / Mass: 1103.334 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) / References: H-261 Oligopeptide |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 255 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 255 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: NEUTRON DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density % sol: 39 % Description: HYDROGEN/DEUTERIUM EXCHANGE BY CAPILLARY VAPOUR DIFFUSION. | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.5 / Details: AS FOR 2ER7, PH 4.5 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: NUCLEAR REACTOR / Site:  ILL ILL  / Beamline: LADI / Wavelength: 2.7-3.6 / Beamline: LADI / Wavelength: 2.7-3.6 | |||||||||
| Detector | Detector: IMAGE PLATE / Date: Nov 1, 1999 | |||||||||
| Radiation | Protocol: LAUE / Monochromatic (M) / Laue (L): L / Scattering type: neutron | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 2.1→20 Å / Num. obs: 13548 / % possible obs: 85 % / Observed criterion σ(I): 2 / Redundancy: 3.4 % / Rmerge(I) obs: 0.075 / Rsym value: 0.075 / Net I/σ(I): 5.4 | |||||||||
| Reflection shell | Resolution: 2.1→2.2 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.119 / Mean I/σ(I) obs: 1.7 / % possible all: 72.6 | |||||||||
| Reflection | *PLUS Lowest resolution: 100 Å / % possible obs: 84.5 % | |||||||||
| Reflection shell | *PLUS % possible obs: 72.6 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.1→20 Å / Num. parameters: 20487 / Num. restraintsaints: 97252 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→20 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 20 Å / % reflection Rfree: 5 % / Rfactor obs: 0.2346 / Rfactor Rfree: 0.2742 / Rfactor Rwork: 0.235 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.331 |
