 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1eji | ||||||
|---|---|---|---|---|---|---|---|
| Title | RECOMBINANT SERINE HYDROXYMETHYLTRANSFERASE (MOUSE) | ||||||
 Components Components | SERINE HYDROXYMETHYLTRANSFERASE | ||||||
 Keywords Keywords | TRANSFERASE / SERINE-GLYCINE CONVERSION / PYRIDOXAL 5'-PHOSPHATE / TETRAHYDROFOLATE / ASYMMETRIC DIMER | ||||||
| Function / homology |  Function and homology information Function and homology informationCarnitine synthesis / Metabolism of folate and pterines / cellular response to tetrahydrofolate / purine nucleobase biosynthetic process / serine binding / L-serine catabolic process / glycine metabolic process / L-serine metabolic process / glycine hydroxymethyltransferase / glycine hydroxymethyltransferase activity ...Carnitine synthesis / Metabolism of folate and pterines / cellular response to tetrahydrofolate / purine nucleobase biosynthetic process / serine binding / L-serine catabolic process / glycine metabolic process / L-serine metabolic process / glycine hydroxymethyltransferase / glycine hydroxymethyltransferase activity / glycine biosynthetic process from L-serine / tetrahydrofolate metabolic process / tetrahydrofolate interconversion / dTMP biosynthetic process / folic acid metabolic process / one-carbon metabolic process / mRNA regulatory element binding translation repressor activity / cellular response to leukemia inhibitory factor / mRNA 5'-UTR binding / pyridoxal phosphate binding / protein homotetramerization / protein homodimerization activity / mitochondrion / nucleoplasm / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Szebenyi, D.M.E. / Liu, X. / Kriksunov, I.A. / Stover, P.J. / Thiel, D.J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2000 Title: Structure of a murine cytoplasmic serine hydroxymethyltransferase quinonoid ternary complex: evidence for asymmetric obligate dimers. Authors: Szebenyi, D.M. / Liu, X. / Kriksunov, I.A. / Stover, P.J. / Thiel, D.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1eji.cif.gz | 343.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1eji.ent.gz | 284.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1eji.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ej/1ejiftp://data.pdbj.org/pub/pdb/validation_reports/ej/1eji | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1cj0S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
|
-Components
| #1: Protein | Mass: 53167.594 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: P50431, glycine hydroxymethyltransferase #2: Chemical | ChemComp-PLG /   Mass: 306.209 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N2O7P Mass: 306.209 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N2O7P#3: Chemical | 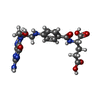  Mass: 473.439 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H23N7O7 Mass: 473.439 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H23N7O7#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 22 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 22 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.33 Å3/Da / Density % sol: 62.3 % Description: RESOLUTION RANGES 3.96-3.84 AND 3.72-3.64 A EXCLUDED FROM PROCESSING BECAUSE OF ICE RINGS. | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.7 Details: 8% PEG 8000, 6% ETHYLENE GLYCOL, 0.1 M HEPES, pH 7.7, VAPOR DIFFUSION, HANGING DROP, temperature 295.0K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F1 / Wavelength: 0.922 / Beamline: F1 / Wavelength: 0.922 |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: May 7, 1999 / Details: MIRROR |
| Radiation | Monochromator: BENT TRIANGULAR SI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.922 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→32.1 Å / Num. obs: 57974 / % possible obs: 92.8 % / Observed criterion σ(I): 0 / Redundancy: 10.6 % / Biso Wilson estimate: 78.8 Å2 / Rsym value: 0.203 / Net I/σ(I): 3.4 |
| Reflection shell | Resolution: 2.9→3.06 Å / Redundancy: 10.9 % / Mean I/σ(I) obs: 0.3 / Rsym value: 2.38 / % possible all: 100 |
| Reflection | *PLUS Num. measured all: 1046890 / Rmerge(I) obs: 0.203 |
| Reflection shell | *PLUS % possible obs: 100 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: RABBIT SHMT (1CJ0) DIMER, WITH 2 PLP COFACTORS. EACH MONOMER WAS MODIFIED BY: OMISSION OF RESIDUES 269-287 (IN THE HUMAN SHMT NUMBERING SCHEME; LABELED 241-250 IN THE 1CJ0 ENTRY), ...Starting model: RABBIT SHMT (1CJ0) DIMER, WITH 2 PLP COFACTORS. EACH MONOMER WAS MODIFIED BY: OMISSION OF RESIDUES 269-287 (IN THE HUMAN SHMT NUMBERING SCHEME; LABELED 241-250 IN THE 1CJ0 ENTRY), CONVERSION OF ALL MET RESIDUES TO SEMET, AND CONVERSION TO ALANINE OF RESIDUES WHICH DIFFER BETWEEN RABBIT AND MOUSE, I.E. RESIDUES 16, 20, 35, 68, 88, 94, 175, 177, 182, 185, 191, 220, 237, 322, 326, 329, 332, 343, 361, 416, 433, 437-439, 441-442, 458, 460, 463, 467, 471, 475, AND 477 IN THE HUMAN SHMT NUMBERING SCHEME. Resolution: 2.9→32.1 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0 / Isotropic thermal model: GROUP / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH & HUBER
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 66.13 Å2 / ksol: 0.347 e/Å3 | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 84.6 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→32.1 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Refine LS restraints NCS |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→3.08 Å / Rfactor Rfree error: 0.012 / Total num. of bins used: 6
| |||||||||||||||||||||||||
| Xplor file |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 10.1 % / Rfactor Rfree: 0.272 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 84.6 Å2 | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.379 / % reflection Rfree: 10.1 % / Rfactor Rwork: 0.371 |
