 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7nn4: Crystal structure of Mycobacterium tuberculosis ArgD with prosthe... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7nn4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Mycobacterium tuberculosis ArgD with prosthetic group pyridoxal 5'-phosphate and 3-hydroxy-2-naphthoic acid. | ||||||
 Components Components | Acetylornithine aminotransferase | ||||||
 Keywords Keywords | TRANSFERASE / ArgD / N-acetylornithine aminotrasferase / AcOAT | ||||||
| Function / homology |  Function and homology information Function and homology informationacetylornithine transaminase / N2-acetyl-L-ornithine:2-oxoglutarate 5-transaminase activity / L-arginine biosynthetic process / pyridoxal phosphate binding / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Mycobacterium tuberculosis H37Rv (bacteria) Mycobacterium tuberculosis H37Rv (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.47 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.47 Å | ||||||
 Authors Authors | Gupta, P. / Mendes, V. / Blundell, T.L. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Comput Struct Biotechnol J / Year: 2021 Title: A fragment-based approach to assess the ligandability of ArgB, ArgC, ArgD and ArgF in the L-arginine biosynthetic pathway of Mycobacterium tuberculosis Authors: Gupta, P. / Thomas, S.E. / Zaidan, S.A. / Pasillas, M.A. / Cory-Wright, J. / Sebastian-Perez, V. / Burgess, A. / Cattermole, E. / Meghir, C. / Abell, C. / Coyne, A.G. / Jacobs, W.R. / ...Authors: Gupta, P. / Thomas, S.E. / Zaidan, S.A. / Pasillas, M.A. / Cory-Wright, J. / Sebastian-Perez, V. / Burgess, A. / Cattermole, E. / Meghir, C. / Abell, C. / Coyne, A.G. / Jacobs, W.R. / Blundell, T.L. / Tiwari, S. / Mendes, V. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7nn4.cif.gz | 322.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7nn4.ent.gz | 256 KB | Display | PDB format |
| PDBx/mmJSON format | 7nn4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nn/7nn4ftp://data.pdbj.org/pub/pdb/validation_reports/nn/7nn4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7nlfC  7nlnC  7nloC  7nlpC  7nlqC  7nlrC  7nlsC  7nltC  7nluC  7nlwC  7nlxC  7nlyC  7nlzC  7nm0C  7nn1C  7nn7C  7nn8C  7nnbC  7nncC  7nnfC  7nniC  7nnqC  7nnrC  7nnvC  7nnwC  7nnyC  7nnzC  7norC  7nosC  7notC  7nouC  7novC  7np0C  7nphC  7npjC  4adbS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41325.035 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis H37Rv (bacteria)Gene: argD, Rv1655, MTCY06H11.20 / Production host: #2: Chemical | ChemComp-BZJ / 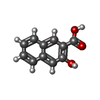  Mass: 188.179 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C11H8O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 188.179 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C11H8O3 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-NO3 / |   Mass: 62.005 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: NO3 Mass: 62.005 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: NO3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1425 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1425 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.85 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 0.1 M Bis-Tris Propane pH 8.5 18% PEG Smear High (PEGs 6K, 8K, 10K) 0.2 M ammonium nitrate 10 mM nickel chloride |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04-1 / Wavelength: 0.9159 Å / Beamline: I04-1 / Wavelength: 0.9159 Å | ||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: May 13, 2019 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9159 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.47→68.47 Å / Num. obs: 265823 / % possible obs: 97.4 % / Redundancy: 4.5 % / CC1/2: 0.999 / Rmerge(I) obs: 0.043 / Rpim(I) all: 0.021 / Rrim(I) all: 0.048 / Net I/σ(I): 16.5 / Num. measured all: 1192624 / Scaling rejects: 5 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4ADB Resolution: 1.47→68.465 Å / SU ML: 0.18 / Cross valid method: THROUGHOUT / σ(F): 1.35 / Phase error: 21.85 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 57.33 Å2 / Biso mean: 24.1684 Å2 / Biso min: 11.75 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.47→68.465 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
|
