 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7lvv | ||||||
|---|---|---|---|---|---|---|---|
| Title | cryoEM structure DrdV-DNA complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/DNA / inhibitor / Complex / endonuclease / methyl transferase / TypeIIL RM system / HYDROLASE / HYDROLASE-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationN-methyltransferase activity / site-specific DNA-methyltransferase (adenine-specific) / site-specific DNA-methyltransferase (adenine-specific) activity / DNA restriction-modification system / endonuclease activity / methylation / DNA binding Similarity search - Function | ||||||
| Biological species |  Deinococcus wulumuqiensis (bacteria) Deinococcus wulumuqiensis (bacteria)synthetic construct (others) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 3.25 Å | ||||||
 Authors Authors | Shen, B.W. / Stoddard, B.L. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Structure / Year: 2021 Title: Coordination of phage genome degradation versus host genome protection by a bifunctional restriction-modification enzyme visualized by CryoEM. Authors: Betty W Shen / Joel D Quispe / Yvette Luyten / Benjamin E McGough / Richard D Morgan / Barry L Stoddard / Abstract: Restriction enzymes that combine methylation and cleavage into a single assemblage and modify one DNA strand are capable of efficient adaptation toward novel targets. However, they must reliably ...Restriction enzymes that combine methylation and cleavage into a single assemblage and modify one DNA strand are capable of efficient adaptation toward novel targets. However, they must reliably cleave invasive DNA and methylate newly replicated unmodified host sites. One possible solution is to enforce a competition between slow methylation at a single unmodified host target, versus faster cleavage that requires multiple unmodified target sites in foreign DNA to be brought together in a reaction synapse. To examine this model, we have determined the catalytic behavior of a bifunctional type IIL restriction-modification enzyme and determined its structure, via cryoelectron microscopy, at several different stages of assembly and coordination with bound DNA targets. The structures demonstrate a mechanism in which an initial dimer is formed between two DNA-bound enzyme molecules, positioning the endonuclease domain from each enzyme against the other's DNA and requiring further additional DNA-bound enzyme molecules to enable cleavage. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7lvv.cif.gz | 497.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7lvv.ent.gz | 381.8 KB | Display | PDB format |
| PDBx/mmJSON format | 7lvv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lv/7lvvftp://data.pdbj.org/pub/pdb/validation_reports/lv/7lvv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  23543MC  7lo5C M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 118256.859 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Deinococcus wulumuqiensis (bacteria) / Gene: drdVRM, DVJ83_12125 / Production host: References: UniProt: A0A345IJ72, site-specific DNA-methyltransferase (adenine-specific) #2: DNA chain | Mass: 8748.646 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #3: DNA chain | Mass: 9085.788 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #4: Chemical | 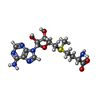  Mass: 398.437 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H22N6O5S / Feature type: SUBJECT OF INVESTIGATION Mass: 398.437 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H22N6O5S / Feature type: SUBJECT OF INVESTIGATION#5: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca / Feature type: SUBJECT OF INVESTIGATION Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: DrdVDNA complex / Type: COMPLEX / Details: dimer of DrdVDNA complex / Entity ID: #1-#3 / Source: RECOMBINANT | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.24 kDa/nm / Experimental value: NO | ||||||||||||
| Source (natural) | Organism: Deinococcus wulumuqiensis (bacteria) | ||||||||||||
| Source (recombinant) | Organism: | ||||||||||||
| Buffer solution | pH: 7.5 / Details: 20 nM HEPES pH7.5 150 NaCl, 2 mM CaCl2 | ||||||||||||
| Buffer component |
| ||||||||||||
| Specimen | Conc.: 0.25 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | ||||||||||||
| Specimen support | Grid material: COPPER / Grid mesh size: 200 divisions/in. / Grid type: Quantifoil R1.2/1.3 | ||||||||||||
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE / Humidity: 95 % / Chamber temperature: 298 K |
- Electron microscopy imaging
Electron microscopy imaging
| Microscopy | Model: TFS GLACIOS |
|---|---|
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: SPOT SCAN FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: SPOT SCAN |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 37000 X / Nominal defocus max: 2300 nm / Nominal defocus min: 800 nm / Cs: 2.7 mm / C2 aperture diameter: 50 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Temperature (max): 70 K / Temperature (min): 70 K |
| Image recording | Average exposure time: 2 sec. / Electron dose: 40 e/Å2 / Film or detector model: FEI CETA (4k x 4k) / Num. of grids imaged: 3 / Num. of real images: 1200 Details: images were collected in movie-mode at 5 frames per second |
| EM imaging optics | Energyfilter slit width: 20 eV |
- Processing
Processing
| EM software |
| |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: NONE | |||||||||||||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 224095 | |||||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | |||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 3.25 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 34554 / Symmetry type: POINT |
