 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7cd9: Crystal Structure of SETDB1 tudor domain in complexed with Compound 6 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7cd9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of SETDB1 tudor domain in complexed with Compound 6 | ||||||
 Components Components | Histone-lysine N-methyltransferase SETDB1 | ||||||
 Keywords Keywords | TRANSFERASE / SETDB1 / Tudor domain / inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationhistone H3K9 dimethyltransferase activity / [histone H3]-N6,N6-dimethyl-lysine9 N-methyltransferase / histone H3K9me2 methyltransferase activity / histone H3K9 trimethyltransferase activity / transposable element silencing by heterochromatin formation / histone H3K9 methyltransferase activity / histone H3K9 monomethyltransferase activity / heterochromatin organization / histone H3K14ac reader activity / histone H3K9me2/3 reader activity ...histone H3K9 dimethyltransferase activity / [histone H3]-N6,N6-dimethyl-lysine9 N-methyltransferase / histone H3K9me2 methyltransferase activity / histone H3K9 trimethyltransferase activity / transposable element silencing by heterochromatin formation / histone H3K9 methyltransferase activity / histone H3K9 monomethyltransferase activity / heterochromatin organization / histone H3K14ac reader activity / histone H3K9me2/3 reader activity / histone H3 methyltransferase activity / DNA methylation-dependent constitutive heterochromatin formation / Regulation of endogenous retroelements by the Human Silencing Hub (HUSH) complex / Regulation of endogenous retroelements by KRAB-ZFP proteins / promoter-specific chromatin binding / PKMTs methylate histone lysines / methylation / negative regulation of gene expression / chromatin binding / chromatin / DNA binding / zinc ion binding / nucleoplasm / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Xiong, L. / Guo, Y. / Mao, X. / Huang, L. / Wu, C. / Yang, S. | ||||||
| Funding support |  China, 1items China, 1items
| ||||||
 Citation Citation | Journal: Angew.Chem.Int.Ed.Engl. / Year: 2021 Title: Structure-Guided Discovery of a Potent and Selective Cell-Active Inhibitor of SETDB1 Tudor Domain. Authors: Guo, Y. / Mao, X. / Xiong, L. / Xia, A. / You, J. / Lin, G. / Wu, C. / Huang, L. / Wang, Y. / Yang, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7cd9.cif.gz | 190.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7cd9.ent.gz | 151.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7cd9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cd/7cd9ftp://data.pdbj.org/pub/pdb/validation_reports/cd/7cd9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7c9nC  7cajC  7cjtC  6bhdS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27613.551 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: SETDB1, ESET, KIAA0067, KMT1E / Production host:  References: UniProt: Q15047, Transferases; Transferring one-carbon groups; Methyltransferases #2: Chemical | 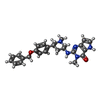  Mass: 443.541 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C26H29N5O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 443.541 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C26H29N5O2 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical |   Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7 / Feature type: SUBJECT OF INVESTIGATION Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 404 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 404 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.29 Å3/Da / Density % sol: 46.19 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, hanging drop Details: 0.2 M Sodium citrate tribasic dihydrate 20% PEG3350 PH 8.3 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL19U1 / Wavelength: 0.97849 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jun 18, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97849 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→50 Å / Num. obs: 64717 / % possible obs: 98.6 % / Redundancy: 6.9 % / Biso Wilson estimate: 12.52 Å2 / CC1/2: 0.996 / Net I/σ(I): 30.257 |
| Reflection shell | Resolution: 1.6→1.63 Å / Num. unique obs: 3196 / CC1/2: 0.996 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6BHD Resolution: 1.6→29.662 Å / SU ML: 0.18 / Cross valid method: THROUGHOUT / σ(F): 1.38 / Phase error: 22.67 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 102.98 Å2 / Biso mean: 21.0988 Å2 / Biso min: 2.23 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.6→29.662 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
|
