 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7au9: Structure of P. aeruginosa PBP3 in complex with a benzoxaborole (... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7au9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of P. aeruginosa PBP3 in complex with a benzoxaborole (Compound 14) | ||||||
 Components Components | Peptidoglycan D,D-transpeptidase FtsI | ||||||
 Keywords Keywords | HYDROLASE / D / D-transpeptidase / boron-binding / divalency | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan glycosyltransferase activity / serine-type D-Ala-D-Ala carboxypeptidase / serine-type D-Ala-D-Ala carboxypeptidase activity / division septum assembly / FtsZ-dependent cytokinesis / penicillin binding / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / proteolysis / plasma membrane Similarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.137 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.137 Å | ||||||
 Authors Authors | Newman, H. / Bellini, B. / Dowson, C.G. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2021 Title: High-Throughput Crystallography Reveals Boron-Containing Inhibitors of a Penicillin-Binding Protein with Di- and Tricovalent Binding Modes. Authors: Newman, H. / Krajnc, A. / Bellini, D. / Eyermann, C.J. / Boyle, G.A. / Paterson, N.G. / McAuley, K.E. / Lesniak, R. / Gangar, M. / von Delft, F. / Brem, J. / Chibale, K. / Schofield, C.J. / Dowson, C.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7au9.cif.gz | 117.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7au9.ent.gz | 84.3 KB | Display | PDB format |
| PDBx/mmJSON format | 7au9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/au/7au9ftp://data.pdbj.org/pub/pdb/validation_reports/au/7au9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7atmC  7atoC  7atwC  7atxC  7au0C  7au1C  7au8C  7aubC  7auhC  6hzrS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 58027.047 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria)Strain: ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1 Gene: ftsI, pbpB, PA4418 / Plasmid: pET47b / Production host: References: UniProt: G3XD46, serine-type D-Ala-D-Ala carboxypeptidase |
|---|---|
| #2: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #3: Chemical | ChemComp-S1E / 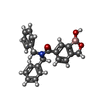  Mass: 357.210 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H20BNO3 / Feature type: SUBJECT OF INVESTIGATION Mass: 357.210 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H20BNO3 / Feature type: SUBJECT OF INVESTIGATION |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 97 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 97 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43.26 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6 Details: 25% (w/v) polyethylene glycol 3,350, 0.1M Bis-Tris propane pH 6 and 1% (w/v) protamine sulphate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I04-1 / Wavelength: 0.91587 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Jan 26, 2019 / Details: Mirrors |
| Radiation | Monochromator: M / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91587 Å / Relative weight: 1 |
| Reflection | Resolution: 2.137→60.446 Å / Num. obs: 21028 / % possible obs: 94.1 % / Redundancy: 8.6 % / CC1/2: 0.999 / Rmerge(I) obs: 0.121 / Rpim(I) all: 0.044 / Net I/σ(I): 12.9 |
| Reflection shell | Resolution: 2.137→2.341 Å / Redundancy: 7.8 % / Rmerge(I) obs: 1.311 / Mean I/σ(I) obs: 1.6 / Num. unique obs: 1052 / CC1/2: 0.582 / Rpim(I) all: 0.499 / % possible all: 67.2 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6HZR Resolution: 2.137→60.446 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.906 / SU B: 8.993 / SU ML: 0.22 / Cross valid method: FREE R-VALUE / ESU R: 0.424 / ESU R Free: 0.29 Details: Hydrogens have been added in their riding positions
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 48.565 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.137→60.446 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.137→2.193 Å
|
