 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6xt7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Tel25 Hybrid Four-quartet G-quadruplex with K+ | |||||||||
 Components Components | DNA (25-MER) | |||||||||
 Keywords Keywords | DNA / G-quadruplex / Hybrid / Four-quartets / lateral loops / propeller loop | |||||||||
| Function / homology | : / SPERMINE / DNA / DNA (> 10) Function and homology information Function and homology information | |||||||||
| Biological species |   Tetrahymena thermophila (eukaryote) Tetrahymena thermophila (eukaryote) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.56 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.56 Å | |||||||||
 Authors Authors | Yatsunyk, L.A. / McCarthy, S.E. | |||||||||
| Funding support |  United States, 1items United States, 1items
| |||||||||
 Citation Citation | Journal: Nucleic Acids Res. / Year: 2022 Title: The first crystal structures of hybrid and parallel four-tetrad intramolecular G-quadruplexes. Authors: Beseiso, D. / Chen, E.V. / McCarthy, S.E. / Martin, K.N. / Gallagher, E.P. / Miao, J. / Yatsunyk, L.A. #1: Journal: Nucleic Acids Res. / Year: 2021Title: Water spines and networks in G-quadruplex structures. Authors: Li, K. / Yatsunyk, L. / Neidle, S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6xt7.cif.gz | 173 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6xt7.ent.gz | 114.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6xt7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xt/6xt7ftp://data.pdbj.org/pub/pdb/validation_reports/xt/6xt7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6w9pC  7jkuC  7ll0C  1jrnS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 7985.084 Da / Num. of mol.: 4 / Source method: obtained synthetically Details: Residues 1007 to 1010 on chain B are split into alternate conformations. Source: (synth.) Tetrahymena thermophila (eukaryote)#2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: K#4: Chemical | 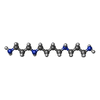  Mass: 202.340 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H26N4 Mass: 202.340 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H26N4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 379 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 379 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.21 Å3/Da / Density % sol: 44.4 % Description: Long rods with a line down the middle. Somewhat opaque. |
|---|---|
| Crystal grow | Temperature: 296 K / Method: vapor diffusion, hanging drop Details: 39% 2-methyl-2,4-pentanediol, 0.165 M potassium chloride, 0.02 M magnesium chloride, 0.04 M sodium cacodylate pH 6.5, 0.012 M spermine tetrahydrochloride PH range: 6.5-7.2 / Temp details: Crystals grown at room temperature. |
-Data collection
| Diffraction | Mean temperature: 196 K / Serial crystal experiment: N | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 24-ID-E / Wavelength: 0.9791 Å | ||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Feb 13, 2020 | ||||||||||||||||||||||||
| Radiation | Monochromator: Cryogenically-cooled single crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9791 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 1.56→93.06 Å / Num. obs: 37569 / % possible obs: 98 % / Redundancy: 6.6 % / Biso Wilson estimate: 27.14 Å2 / Rmerge(I) obs: 0.071 / Rpim(I) all: 0.03 / Rrim(I) all: 0.082 / Net I/σ(I): 15.4 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1JRN Resolution: 1.56→49.38 Å / SU ML: 0.1486 / Cross valid method: FREE R-VALUE / σ(F): 1.37 / Phase error: 25.689 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39.73 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.56→49.38 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -8.58246456259 Å / Origin y: -27.3649458929 Å / Origin z: -10.9788063564 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
