 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6xfu | ||||||
|---|---|---|---|---|---|---|---|
| Title | PmtCD peptide exporter basket domain | ||||||
 Components Components | ABC transporter ATP-binding protein | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / ABC transporter / ABC exporter / Peptide transort | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.4 Å | ||||||
 Authors Authors | Zeytuni, N. / Strynadka, N.C.J. / Alexander, J.A.N. | ||||||
| Funding support |  Canada, 1items Canada, 1items
| ||||||
 Citation Citation | Journal: Sci Adv / Year: 2020 Title: Structural insight into the ATP-driven exporter of virulent peptide toxins. Authors: N Zeytuni / S W Dickey / J Hu / H T Chou / L J Worrall / J A N Alexander / M L Carlson / M Nosella / F Duong / Z Yu / M Otto / N C J Strynadka /  Abstract: is a major human pathogen that has acquired alarming broad-spectrum antibiotic resistance. One group of secreted toxins with key roles during infection is the phenol-soluble modulins (PSMs). PSMs ... is a major human pathogen that has acquired alarming broad-spectrum antibiotic resistance. One group of secreted toxins with key roles during infection is the phenol-soluble modulins (PSMs). PSMs are amphipathic, membrane-destructive cytolytic peptides that are exported to the host-cell environment by a designated adenosine 5'-triphosphate (ATP)-binding cassette (ABC) transporter, the PSM transporter (PmtABCD). Here, we demonstrate that the minimal Pmt unit necessary for PSM export is PmtCD and provide its first atomic characterization by single-particle cryo-EM and x-ray crystallography. We have captured the transporter in the ATP-bound state at near atomic resolution, revealing a type II ABC exporter fold, with an additional cytosolic domain. Comparison to a lower-resolution nucleotide-free map displaying an "open" conformation and putative hydrophobic inner chamber of a size able to accommodate the binding of two PSM peptides provides mechanistic insight and sets the foundation for therapeutic design. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6xfu.cif.gz | 48.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6xfu.ent.gz | 33.1 KB | Display | PDB format |
| PDBx/mmJSON format | 6xfu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xf/6xfuftp://data.pdbj.org/pub/pdb/validation_reports/xf/6xfu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6u2dSC  6xjhC  6xjiC  6u4h S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 8166.194 Da / Num. of mol.: 2 / Fragment: basket domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria)Gene: ybhF_2, pmtC, ybhF_1, ybhF_3, BN1321_320024, BTN44_02050, C7P97_04805, CSC83_02360, CSC87_13000, EP54_10395, EQ90_13985, ER624_12880, ERS072840_01916, HMPREF3211_02244, NCTC10654_02103, ...Gene: ybhF_2, pmtC, ybhF_1, ybhF_3, BN1321_320024, BTN44_02050, C7P97_04805, CSC83_02360, CSC87_13000, EP54_10395, EQ90_13985, ER624_12880, ERS072840_01916, HMPREF3211_02244, NCTC10654_02103, NCTC13131_01274, NCTC7878_02640, NCTC7988_02070, RK64_10625 Production host: #2: Chemical | 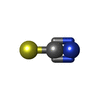  Mass: 58.082 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CNS / Feature type: SUBJECT OF INVESTIGATION Mass: 58.082 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CNS / Feature type: SUBJECT OF INVESTIGATION#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 168 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 168 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.09 Å3/Da / Density % sol: 41.03 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7.4 Details: 0.2 M sodium thiocyanate and 20% Poly Ethylene Glycol (PEG) 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 23-ID-B / Wavelength: 1.033 Å | ||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: May 30, 2020 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.033 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.4→45.37 Å / Num. obs: 27091 / % possible obs: 98.5 % / Redundancy: 11.6 % / CC1/2: 0.998 / Rmerge(I) obs: 0.098 / Rpim(I) all: 0.029 / Rrim(I) all: 0.103 / Net I/σ(I): 14.7 / Num. measured all: 314139 / Scaling rejects: 37 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6U2D Resolution: 1.4→40.46 Å / Cor.coef. Fo:Fc: 0.974 / Cor.coef. Fo:Fc free: 0.965 / WRfactor Rfree: 0.2086 / WRfactor Rwork: 0.1699 / FOM work R set: 0.8736 / SU B: 0.992 / SU ML: 0.039 / SU R Cruickshank DPI: 0.0598 / SU Rfree: 0.0639 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.06 / ESU R Free: 0.064 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 54.13 Å2 / Biso mean: 18.273 Å2 / Biso min: 10.54 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.4→40.46 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.4→1.436 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
