 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-20640: PmtCD peptide toxin ABC exporter in nucleotide free (apo) conformation -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-20640 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
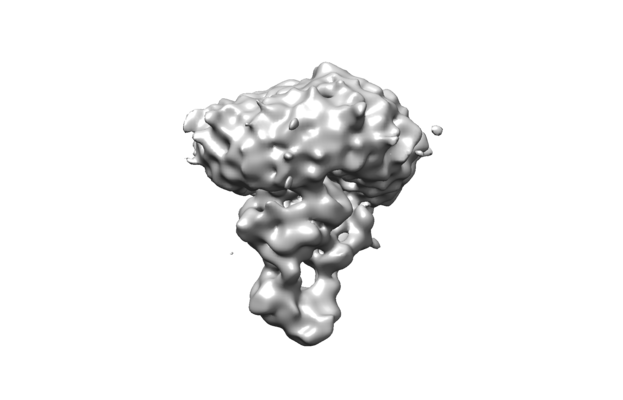
| Title | PmtCD peptide toxin ABC exporter in nucleotide free (apo) conformation | |||||||||
 Map data Map data | PmtCD toxin peptide ABC exporter in nucleotide free (apo) conformation | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | |||||||||
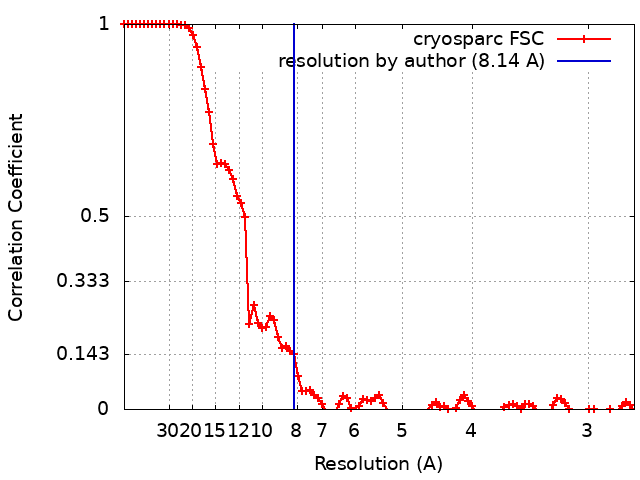
| Method | single particle reconstruction / cryo EM / Resolution: 8.14 Å | |||||||||
 Authors Authors | Zeytuni N / Strynadka NCJ / Yu Z / Chou HT | |||||||||
| Funding support |  Canada, 1 items Canada, 1 items
| |||||||||
 Citation Citation | Journal: Sci Adv / Year: 2020 Title: Structural insight into the ATP-driven exporter of virulent peptide toxins. Authors: N Zeytuni / S W Dickey / J Hu / H T Chou / L J Worrall / J A N Alexander / M L Carlson / M Nosella / F Duong / Z Yu / M Otto / N C J Strynadka /  Abstract: is a major human pathogen that has acquired alarming broad-spectrum antibiotic resistance. One group of secreted toxins with key roles during infection is the phenol-soluble modulins (PSMs). PSMs ... is a major human pathogen that has acquired alarming broad-spectrum antibiotic resistance. One group of secreted toxins with key roles during infection is the phenol-soluble modulins (PSMs). PSMs are amphipathic, membrane-destructive cytolytic peptides that are exported to the host-cell environment by a designated adenosine 5'-triphosphate (ATP)-binding cassette (ABC) transporter, the PSM transporter (PmtABCD). Here, we demonstrate that the minimal Pmt unit necessary for PSM export is PmtCD and provide its first atomic characterization by single-particle cryo-EM and x-ray crystallography. We have captured the transporter in the ATP-bound state at near atomic resolution, revealing a type II ABC exporter fold, with an additional cytosolic domain. Comparison to a lower-resolution nucleotide-free map displaying an "open" conformation and putative hydrophobic inner chamber of a size able to accommodate the binding of two PSM peptides provides mechanistic insight and sets the foundation for therapeutic design. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_20640.map.gz | 59.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-20640-v30.xmlemd-20640.xml | 12.1 KB 12.1 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_20640_fsc.xml | 9.3 KB | Display | FSC data file |
| Images |  emd_20640.png emd_20640.png | 61 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-20640ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20640 http://ftp.pdbj.org/pub/emdb/structures/EMD-20640ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20640 | HTTPS FTP |
-Related structure data
| Related structure data |  6u2dC  6xfuC  6xjhC  6xjiC  20639 C: citing same article ( |
|---|---|
| Similar structure data |











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_20640.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | PmtCD toxin peptide ABC exporter in nucleotide free (apo) conformation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.35 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : PmtCD
| Entire | Name: PmtCD |
|---|---|
| Components |
|
-Supramolecule #1: PmtCD
| Supramolecule | Name: PmtCD / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all / Details: PmtCD ABC exporter |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (bacteria) |
| Molecular weight | Experimental: 120 KDa |
-Macromolecule #1: PmtD
| Macromolecule | Name: PmtD / type: protein_or_peptide / ID: 1 / Enantiomer: DEXTRO |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (bacteria) |
| Sequence | String: MRILNLVKYD FYSIFKSPLT YLAILVVSSL IATQSILMAN SMDNPKHIIV YGSVFAAAKW LLLIIGLMF VVKTITRDFS QGTIQLYMSK VKTRVGYIIS KTISIILISI LFALIHYVIL I VVQASSNG KNLAFSKYVD NLWFFLIFLL FFGLFLFLIT LASQKTAMIF ...String: MRILNLVKYD FYSIFKSPLT YLAILVVSSL IATQSILMAN SMDNPKHIIV YGSVFAAAKW LLLIIGLMF VVKTITRDFS QGTIQLYMSK VKTRVGYIIS KTISIILISI LFALIHYVIL I VVQASSNG KNLAFSKYVD NLWFFLIFLL FFGLFLFLIT LASQKTAMIF SLGVFLVLIV PF IKPFITF IPRYGEKVLD AFDYIPFAYL TDKMISSNFD FSNWQWVISL GSIVIFFILN ILY VAKKDI |
-Macromolecule #2: PmtC
| Macromolecule | Name: PmtC / type: protein_or_peptide / ID: 2 / Enantiomer: DEXTRO |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (bacteria) |
| Sequence | String: MKLEHITKKY GSNVVLNDID FDFGDSRIVG LIGKNGVGKT TVMKVMNGNI IKFDGKVDID NADNIGFLI EHPKLYDNKS GLYNLKLFAQ VLGKGFDKAY TDKIIDAFGM RPYIKKKVKK Y SMGMKQKL AIAVSLMNKP KFLILDEPTN GMDPDGSIDV LTTIKSLVNE ...String: MKLEHITKKY GSNVVLNDID FDFGDSRIVG LIGKNGVGKT TVMKVMNGNI IKFDGKVDID NADNIGFLI EHPKLYDNKS GLYNLKLFAQ VLGKGFDKAY TDKIIDAFGM RPYIKKKVKK Y SMGMKQKL AIAVSLMNKP KFLILDEPTN GMDPDGSIDV LTTIKSLVNE LDMRILISSH KL EDIELIC DRAVFLRDGH FVQDVNMEEG VASDTTIVTV DHKDFDRTEK YLAEHFQLQN VDK ADGHLM INAQKNYQVI LKALSELDIY PKYIETRKSS LRDTYFNINQ RGDK |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.4 Component:
Details: Solution made fresh from concentrated stocks and filtered | |||||||||
| Grid | Support film - Material: CARBON / Support film - topology: HOLEY / Details: unspecified | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK I |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Average electron dose: 1.45 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Protocol: AB INITIO MODEL |
|---|
