 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6s1u: Crystal structure of dimeric M-PMV protease C7A/D26N/C106A mutant... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6s1u | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of dimeric M-PMV protease C7A/D26N/C106A mutant in complex with inhibitor | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / Mason-Pfizer Monkey Virus / M-PMV / retrovirus / retropepsin / aspartic protease / dimerization / inhibitor / flap structure | ||||||
| Function / homology |  Function and homology information Function and homology informationdUTP diphosphatase / dUTP diphosphatase activity / Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / ribonuclease H / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / RNA-directed DNA polymerase activity ...dUTP diphosphatase / dUTP diphosphatase activity / Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / ribonuclease H / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / structural constituent of virion / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / viral translational frameshifting / symbiont entry into host cell / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Mason-Pfizer monkey virus Mason-Pfizer monkey virussynthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Wosicki, S. / Gilski, M. / Jaskolski, M. / Zabranska, H. / Pichova, I. | ||||||
| Funding support |  Czech Republic, 1items Czech Republic, 1items
| ||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2019 Title: Comparison of a retroviral protease in monomeric and dimeric states. Authors: Wosicki, S. / Gilski, M. / Zabranska, H. / Pichova, I. / Jaskolski, M. #1: Journal: Nat.Struct.Mol.Biol. / Year: 2011Title: Crystal structure of a monomeric retroviral protease solved by protein folding game players. Authors: Khatib, F. / DiMaio, F. / Cooper, S. / Kazmierczyk, M. / Gilski, M. / Krzywda, S. / Zabranska, H. / Pichova, I. / Thompson, J. / Popovic, Z. / Jaskolski, M. / Baker, D. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2011 Title: High-resolution structure of a retroviral protease folded as a monomer. Authors: Gilski, M. / Kazmierczyk, M. / Krzywda, S. / Zabranska, H. / Cooper, S. / Popovic, Z. / Khatib, F. / DiMaio, F. / Thompson, J. / Baker, D. / Pichova, I. / Jaskolski, M. #3: Journal: Nature / Year: 1989 Title: Crystal structure of a retroviral protease proves relationship to aspartic protease family. Authors: Miller, M. / Jaskolski, M. / Rao, J.K. / Leis, J. / Wlodawer, A. #4: Journal: Science / Year: 1989Title: Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Authors: Wlodawer, A. / Miller, M. / Jaskolski, M. / Sathyanarayana, B.K. / Baldwin, E. / Weber, I.T. / Selk, L.M. / Clawson, L. / Schneider, J. / Kent, S.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6s1u.cif.gz | 106.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6s1u.ent.gz | 81 KB | Display | PDB format |
| PDBx/mmJSON format | 6s1u.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/s1/6s1uftp://data.pdbj.org/pub/pdb/validation_reports/s1/6s1u | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6s1vC  6s1wC  3sqfS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Experimental dataset #1 | Data reference: 10.18150/repod.0005795 / Data set type: diffraction image data / Metadata reference: 10.18150/repod.0005795 |


















-Links
 PDBj
PDBj


- Assembly
Assembly
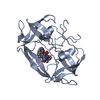
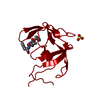
| Deposited unit | 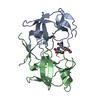
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: _ / Ens-ID: 1 / Beg auth comp-ID: TRP / Beg label comp-ID: TRP / End auth comp-ID: PRO / End label comp-ID: PRO / Refine code: _ / Auth seq-ID: 1 - 108 / Label seq-ID: 1 - 108
|
-Components
| #1: Protein | Mass: 12899.834 Da / Num. of mol.: 2 / Mutation: C7A, D26N, C106A; ENGINEERED MUTATION Source method: isolated from a genetically manipulated source Details: Gaps in the sequence indicate residues that were not modeled because of poor electron density. Source: (gene. exp.) Mason-Pfizer monkey virus / Gene: gag-pro-pol / Plasmid: pBPS13ATGProduction host:  References: UniProt: P07572, dUTP diphosphatase, Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases, RNA-directed DNA polymerase, DNA-directed DNA polymerase, ribonuclease H, ...References: UniProt: P07572, dUTP diphosphatase, Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases, RNA-directed DNA polymerase, DNA-directed DNA polymerase, ribonuclease H, Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases, Hydrolases; Acting on ester bonds #2: Protein/peptide | | Mass: 886.065 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: In the standard definition used by Refmac, the Cgamma atom of the PSA residue is labeled as CA. In the PDB Validation Report this label is interpreted as Calpha causing geometrical alerts. ...Details: In the standard definition used by Refmac, the Cgamma atom of the PSA residue is labeled as CA. In the PDB Validation Report this label is interpreted as Calpha causing geometrical alerts. These alerts are false and should be ignored. Source: (synth.) synthetic construct (others) #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 149 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 149 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.365 Å3/Da / Density % sol: 47.99 % / Description: plate |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: Protein solution: 6.8 mg/mL protein with 1.2-fold molar excess (relative to dimeric protein) of Pro-0A1-Val-PSA-Ala-Met-Thr (inhibitor), 10 mM Tris buffer pH 7.4; Reservoir solution: 0.1 M ...Details: Protein solution: 6.8 mg/mL protein with 1.2-fold molar excess (relative to dimeric protein) of Pro-0A1-Val-PSA-Ala-Met-Thr (inhibitor), 10 mM Tris buffer pH 7.4; Reservoir solution: 0.1 M imidazole buffer, 1.2 M sodium acetate; |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.2 / Wavelength: 0.91841 Å / Beamline: 14.2 / Wavelength: 0.91841 Å |
| Detector | Type: RAYONIX MX-225 / Detector: CCD / Date: Apr 22, 2012 |
| Radiation | Monochromator: Double Crystal Monochromator, Si-111 crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91841 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→48.3 Å / Num. obs: 20007 / % possible obs: 99.1 % / Redundancy: 4.02 % / Biso Wilson estimate: 29.7 Å2 / CC1/2: 0.997 / Rmerge(I) obs: 0.108 / Rrim(I) all: 0.124 / Net I/σ(I): 11.49 |
| Reflection shell | Resolution: 1.9→2.02 Å / Redundancy: 3.67 % / Rmerge(I) obs: 0.98 / Mean I/σ(I) obs: 1.53 / Num. unique obs: 3082 / CC1/2: 0.969 / Rrim(I) all: 1.146 / % possible all: 95.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3sqf, chain A Resolution: 1.9→48.3 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.935 / SU B: 8.787 / SU ML: 0.12 / Cross valid method: FREE R-VALUE / ESU R: 0.151 / ESU R Free: 0.144 / Details: HYDROGEN ATOMS WERE ADDED AT RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.38 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.9→48.3 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|