 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6fiv: STRUCTURAL STUDIES OF HIV AND FIV PROTEASES COMPLEXED WITH AN EFF... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6fiv | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURAL STUDIES OF HIV AND FIV PROTEASES COMPLEXED WITH AN EFFICIENT INHIBITOR OF FIV PR | ||||||
 Components Components | RETROPEPSIN | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / FIV PROTEASE / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationdUTP catabolic process / dUMP biosynthetic process / dUTP diphosphatase / dUTP diphosphatase activity / Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA ...dUTP catabolic process / dUMP biosynthetic process / dUTP diphosphatase / dUTP diphosphatase activity / Hydrolases; Acting on peptide bonds (peptidases); Aspartic endopeptidases / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / DNA recombination / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / symbiont entry into host cell / magnesium ion binding / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Feline immunodeficiency virus Feline immunodeficiency virus | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 1.9 Å X-RAY DIFFRACTION / OTHER / Resolution: 1.9 Å | ||||||
 Authors Authors | Li, M. / Lee, T. / Morris, G. / Laco, G. / Wong, C. / Olson, A. / Elder, J. / Wlodawer, A. / Gustchina, A. | ||||||
 Citation Citation | Journal: Proteins / Year: 2000 Title: Structural studies of FIV and HIV-1 proteases complexed with an efficient inhibitor of FIV protease Authors: Li, M. / Morris, G.M. / Lee, T. / Laco, G.S. / Wong, C.H. / Olson, A.J. / Elder, J.H. / Wlodawer, A. / Gustchina, A. #1: Journal: Nat.Struct.Biol. / Year: 1995Title: Structural Studies of HIV and Fiv Proteases Complexed with an Efficient Inhibitor of Fiv Pr Authors: Wlodawer, A. / Gustchina, A. / Reshetnikova, L. / Lubkowski, J. / Zdanov, A. / Hui, K. / Angleton, E. / Farmerie, W. / Goodenow, M. / Bhatt, D. / Zhang, L. / Dunn, B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6fiv.cif.gz | 40.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6fiv.ent.gz | 26.1 KB | Display | PDB format |
| PDBx/mmJSON format | 6fiv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fi/6fivftp://data.pdbj.org/pub/pdb/validation_reports/fi/6fiv | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 12813.828 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: COMPLEXED WITH TL-3-093 / Source: (gene. exp.) Feline immunodeficiency virus / Genus: Lentivirus / Strain: (isolate Petaluma) / Production host:  |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-3TL / 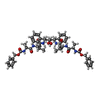  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 909.077 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C50H64N6O10 Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 909.077 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C50H64N6O10References: N-[(benzyloxy)carbonyl]-L-alanyl-N-[(1R)-1-benzyl-2-oxoethyl]-L-valinamide |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 76 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 76 / Source method: isolated from a natural source / Formula: H2O |
| Nonpolymer details | THE INHIBITOR IS A C2 SYMMETRIC HIV PROTEASE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.4 % | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5 Details: SAMPLE: 3.4MG/ML Q99V IN 25MM IMIDAZOL BUFFER AT PH 7.0 WITH 1MM EDTA AND 10MM DTT. WELL SOLUTION: 1.6M AMMONIUM SULFATE IN SODIUM ACETATE BUFFER AT PH 4.5 MIXING SAMPLE AND WELL SOLLUTION 1:1, pH 5.0 PH range: 4.5-7.0 | |||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7 / Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: SIEMENS HI-STAR / Detector: AREA DETECTOR / Date: Nov 1, 1997 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→20 Å / Num. obs: 8437 / % possible obs: 3.1 % / Observed criterion σ(I): 0 / Redundancy: 6.3 % / Rmerge(I) obs: 0.078 |
| Reflection shell | Resolution: 1.9→1.97 Å / Rmerge(I) obs: 0.372 / Mean I/σ(I) obs: 1.98 / % possible all: 75 |
| Reflection | *PLUS % possible obs: 93.1 % |
| Reflection shell | *PLUS % possible obs: 75 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 1.9→10 Å / Num. parameters: 4069 / Num. restraintsaints: 3813 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC SCALING APPLIED BY METHOD PF PARKIN, MOEZZI & HOPE, J.APPL.CRYST.28(1995)53-56
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 2 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 991 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.155 / Rfactor Rfree: 0.266 / Rfactor Rwork: 0.157 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 25.39 Å2 | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
