Entry Database : PDB / ID : 6q5qTitle Crystal structure of a CC-Hex mutant that forms an antiparallel four-helix coiled coil CC-Hex-KgEb CC-Hex-KgEb Keywords / / / / Function / homology Biological species synthetic construct (others) Method / / / Resolution : 1.08 Å Authors Zaccai, N.R. / Rhys, G.G. / Wood, C.W. / Beesley, J.L. / Brady, R.L. / Woolfson, D.N. Funding support Organization Grant number Country Biotechnology and Biological Sciences Research Council BB/J014400/1 Engineering and Physical Sciences Research Council EP/G036764/1 European Research Council 340764 Biotechnology and Biological Sciences Research Council BB/R00661X/1 Engineering and Physical Sciences Research Council EP/K03927X/1
Journal : J.Am.Chem.Soc. / Year : 2019Title : Navigating the Structural Landscape of De Novo alpha-Helical Bundles.Authors : Rhys, G.G. / Wood, C.W. / Beesley, J.L. / Zaccai, N.R. / Burton, A.J. / Brady, R.L. / Thomson, A.R. / Woolfson, D.N. History Deposition Dec 9, 2018 Deposition site / Processing site Revision 1.0 May 22, 2019 Provider / Type Revision 1.1 Jun 19, 2019 Group / Database references / Category / citation_author / pdbx_database_procItem _citation.journal_volume / _citation.page_first ... _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.title / _citation_author.identifier_ORCID / _citation_author.name Revision 1.2 Nov 6, 2024 Group / Database references / Structure summaryCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_entry_details / pdbx_modification_feature Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United Kingdom,
United Kingdom,  Belgium, 5items
Belgium, 5items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

















 PDBj
PDBj

 Assembly
Assembly

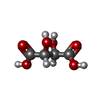
 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6
Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 18.015 Da / Num. of mol.: 34 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 34 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing