 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6lzn | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Thermolysin | |||||||||
 Components Components | Thermolysin | |||||||||
 Keywords Keywords | HYDROLASE / Thermolysin / metalloproteinase | |||||||||
| Function / homology |  Function and homology information Function and homology informationthermolysin / metalloendopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | |||||||||
 Authors Authors | Nam, K.H. | |||||||||
| Funding support |  Korea, Republic Of, 2items Korea, Republic Of, 2items
| |||||||||
 Citation Citation | Journal: J.Inorg.Biochem. / Year: 2021 Title: Structural analysis of metal chelation of the metalloproteinase thermolysin by 1,10-phenanthroline. Authors: Nam, K.H. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6lzn.cif.gz | 85.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6lzn.ent.gz | 61.7 KB | Display | PDB format |
| PDBx/mmJSON format | 6lzn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lz/6lznftp://data.pdbj.org/pub/pdb/validation_reports/lz/6lzn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6lzoC  6ig7S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 34360.336 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|
-Non-polymers , 7 types, 275 molecules 

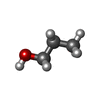


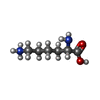

| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #3: Chemical |  Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-POL / |  Mass: 60.095 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O Mass: 60.095 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O#5: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#6: Chemical | ChemComp-ILE / |  Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO2 Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO2#7: Chemical | ChemComp-LYS / |  Type: L-peptide linking / Mass: 147.195 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N2O2 Type: L-peptide linking / Mass: 147.195 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N2O2#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 267 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 47.02 % |
|---|---|
| Crystal grow | Temperature: 294.15 K / Method: vapor diffusion, hanging drop / pH: 8.5 / Details: Tris-HCl, glycerol, Ammonium sulfate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PAL/PLS / Beamline: 7A (6B, 6C1) / Wavelength: 0.9795 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: May 27, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→20 Å / Num. obs: 51659 / % possible obs: 97.9 % / Redundancy: 9.7 % / Rmerge(I) obs: 0.113 / Rpim(I) all: 0.036 / Rrim(I) all: 0.118 / Net I/σ(I): 40.72 |
| Reflection shell | Resolution: 1.5→1.53 Å / Rmerge(I) obs: 0.339 / Mean I/σ(I) obs: 3.25 / Num. unique obs: 2137 / Rpim(I) all: 0.175 / Rrim(I) all: 0.384 / % possible all: 82.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6IG7 Resolution: 1.5→19.83 Å / Cor.coef. Fo:Fc: 0.971 / Cor.coef. Fo:Fc free: 0.964 / SU B: 1.113 / SU ML: 0.041 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.064 / ESU R Free: 0.065 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 68 Å2 / Biso mean: 15.744 Å2 / Biso min: 8.61 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.5→19.83 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.538 Å / Rfactor Rfree error: 0
|
