 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6iy4: Crystal structure of a psychrophilic marine protease MP inhibitor -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6iy4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a psychrophilic marine protease MP inhibitor | ||||||
 Components Components | LupI | ||||||
 Keywords Keywords | HYDROLASE INHIBITOR / psychrophilic marine protease inhibitor | ||||||
| Function / homology | Protease inhibitor / Alkaline proteinase inhibitor/ Outer membrane lipoprotein Omp19 / Protease inhibitor Inh / Protease inhibitor, beta-barrel domain / metalloendopeptidase inhibitor activity / periplasmic space / LupI Function and homology information Function and homology information | ||||||
| Biological species |  Flavobacterium sp. YS-80-122 (bacteria) Flavobacterium sp. YS-80-122 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.59 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.59 Å | ||||||
 Authors Authors | Hao, J.H. / Zhang, L.H. | ||||||
| Funding support |  China, 1items China, 1items
| ||||||
 Citation Citation | Journal: To be published Title: Crystal structure of a psychrophilic marine protease MP inhibitor Authors: Hao, J.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6iy4.cif.gz | 30.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6iy4.ent.gz | 18.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6iy4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6iy4_validation.pdf.gz | 420.5 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6iy4_full_validation.pdf.gz | 420.4 KB | Display | |
| Data in XML | 6iy4_validation.xml.gz | 5.4 KB | Display | |
| Data in CIF | 6iy4_validation.cif.gz | 6.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/iy/6iy4ftp://data.pdbj.org/pub/pdb/validation_reports/iy/6iy4 | HTTPS FTP |
-Related structure data
| Related structure data |  6ixxS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 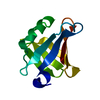
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 10104.364 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Flavobacterium sp. YS-80-122 (bacteria) / Plasmid details: yellow sea in china / References: UniProt: G3MEU6 |
|---|---|
| #2: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 13 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 13 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | Author states that the sequence G3MEU6 may have errors in sequencing, and this time the sequence is ...Author states that the sequence G3MEU6 may have errors in sequencing, and this time the sequence is re-sequence verified to be Leu. |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.07 Å3/Da / Density % sol: 40.57 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: evaporation / pH: 6.5 / Details: 0.1M NaCl, 0.1M BIS-TRIS pH 6.5, 1.5M (NH4)2SO4 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL17U / Wavelength: 0.97915 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jun 15, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97915 Å / Relative weight: 1 |
| Reflection | Resolution: 1.59→50 Å / Num. obs: 11707 / % possible obs: 99.7 % / Redundancy: 5.4 % / CC1/2: 0.991 / Rmerge(I) obs: 0.213 / Rpim(I) all: 0.088 / Net I/σ(I): 11 |
| Reflection shell | Resolution: 1.59→1.64 Å / Rmerge(I) obs: 0.654 / Num. unique obs: 5628 / CC1/2: 0.869 / Rpim(I) all: 0.277 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6IXX Resolution: 1.59→31.31 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.956 / SU B: 1.784 / SU ML: 0.063 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.1 / ESU R Free: 0.09 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 67.91 Å2 / Biso mean: 25.274 Å2 / Biso min: 16.75 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.59→31.31 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.586→1.627 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
