 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6gzd | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Human CSNK1A1 with A86 | ||||||
 Components Components | Casein kinase I isoform alpha | ||||||
 Keywords Keywords | TRANSFERASE / CKIa / Casein kinase / Kinase inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationActivation of SMO / intermediate filament cytoskeleton organization / negative regulation of NLRP3 inflammasome complex assembly / cellular response to nutrient / beta-catenin destruction complex / APC truncation mutants have impaired AXIN binding / AXIN missense mutants destabilize the destruction complex / Truncations of AMER1 destabilize the destruction complex / Beta-catenin phosphorylation cascade / Signaling by GSK3beta mutants ...Activation of SMO / intermediate filament cytoskeleton organization / negative regulation of NLRP3 inflammasome complex assembly / cellular response to nutrient / beta-catenin destruction complex / APC truncation mutants have impaired AXIN binding / AXIN missense mutants destabilize the destruction complex / Truncations of AMER1 destabilize the destruction complex / Beta-catenin phosphorylation cascade / Signaling by GSK3beta mutants / CTNNB1 S33 mutants aren't phosphorylated / CTNNB1 S37 mutants aren't phosphorylated / CTNNB1 S45 mutants aren't phosphorylated / CTNNB1 T41 mutants aren't phosphorylated / Disassembly of the destruction complex and recruitment of AXIN to the membrane / Maturation of nucleoprotein / positive regulation of Rho protein signal transduction / Golgi organization / positive regulation of TORC1 signaling / Degradation of GLI2 by the proteasome / GLI3 is processed to GLI3R by the proteasome / negative regulation of canonical Wnt signaling pathway / Degradation of beta-catenin by the destruction complex / kinetochore / spindle / Wnt signaling pathway / positive regulation of proteasomal ubiquitin-dependent protein catabolic process / proteasome-mediated ubiquitin-dependent protein catabolic process / cell surface receptor signaling pathway / protein phosphorylation / non-specific serine/threonine protein kinase / protein kinase activity / viral protein processing / nuclear speck / cilium / ciliary basal body / cell division / protein serine kinase activity / protein serine/threonine kinase activity / centrosome / signal transduction / ATP binding / nucleus / membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.28 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.28 Å | ||||||
 Authors Authors | Ben-Neriah, Y. / Venkatachalam, A. / Minzel, W. / Fink, A. / Snir-Alkalay, I. / Vacca, J. | ||||||
 Citation Citation | Journal: Cell / Year: 2018 Title: Small Molecules Co-targeting CKI alpha and the Transcriptional Kinases CDK7/9 Control AML in Preclinical Models. Authors: Minzel, W. / Venkatachalam, A. / Fink, A. / Hung, E. / Brachya, G. / Burstain, I. / Shaham, M. / Rivlin, A. / Omer, I. / Zinger, A. / Elias, S. / Winter, E. / Erdman, P.E. / Sullivan, R.W. / ...Authors: Minzel, W. / Venkatachalam, A. / Fink, A. / Hung, E. / Brachya, G. / Burstain, I. / Shaham, M. / Rivlin, A. / Omer, I. / Zinger, A. / Elias, S. / Winter, E. / Erdman, P.E. / Sullivan, R.W. / Fung, L. / Mercurio, F. / Li, D. / Vacca, J. / Kaushansky, N. / Shlush, L. / Oren, M. / Levine, R. / Pikarsky, E. / Snir-Alkalay, I. / Ben-Neriah, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6gzd.cif.gz | 91 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6gzd.ent.gz | 65.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6gzd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6gzd_validation.pdf.gz | 1.4 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6gzd_full_validation.pdf.gz | 1.4 MB | Display | |
| Data in XML | 6gzd_validation.xml.gz | 17.4 KB | Display | |
| Data in CIF | 6gzd_validation.cif.gz | 24.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gz/6gzdftp://data.pdbj.org/pub/pdb/validation_reports/gz/6gzd | HTTPS FTP |
-Related structure data
| Related structure data |  6gzhC  6gzmC  5fqdS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 43075.164 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CSNK1A1 / Production host:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm)References: UniProt: P48729, non-specific serine/threonine protein kinase |
|---|
-Non-polymers , 7 types, 208 molecules 




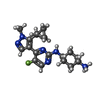

| #2: Chemical |  Mass: 78.133 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#3: Chemical |  Mass: 150.173 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14O4#4: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 5 / Source method: isolated from a natural source / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 5 / Source method: isolated from a natural source / Formula: C2H6O2#5: Chemical | ChemComp-PEG /  Mass: 106.120 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C4H10O3#6: Chemical | ChemComp-TCE / |  Mass: 250.186 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H15O6P Mass: 250.186 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H15O6P#7: Chemical | ChemComp-LCI / [ |  Mass: 345.438 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H26FN6 Mass: 345.438 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H26FN6#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 190 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.02 Å3/Da / Density % sol: 59.23 % |
|---|---|
| Crystal grow | Temperature: 277.15 K / Method: vapor diffusion, sitting drop / pH: 8 Details: CSNK1A1 at a concentration of 5.8 mg/ml (50 mM Tris-HCl, 300 mM NaCl, 0.25 mM TCEP, pH 8.0) was pre-incubated with 0.7 mM (4.7-fold molar excess) of Mg2+-ATP (120 mM in UPW) for 1 h. 0.1 uL ...Details: CSNK1A1 at a concentration of 5.8 mg/ml (50 mM Tris-HCl, 300 mM NaCl, 0.25 mM TCEP, pH 8.0) was pre-incubated with 0.7 mM (4.7-fold molar excess) of Mg2+-ATP (120 mM in UPW) for 1 h. 0.1 uL of the protein solution was then mixed with 0.1 uL of reservoir solution (0.10 M MES/NaOH pH 6.8, 10.0% 2-propanol, 26.0% PEG400) and equilibrated at 4 C over 0.06mL of reservoir solution. Well diffracting crystals appeared within 4 days and grew to full size over 23 days. |
-Data collection
| Diffraction | Mean temperature: 100.5 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: MASSIF-1 / Wavelength: 0.966 Å / Beamline: MASSIF-1 / Wavelength: 0.966 Å |
| Detector | Type: DECTRIS PILATUS3 2M / Detector: PIXEL / Date: Jun 23, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.966 Å / Relative weight: 1 |
| Reflection | Resolution: 2.28→29.31 Å / Num. obs: 22991 / % possible obs: 98.1 % / Redundancy: 4 % / Rrim(I) all: 0.07 / Net I/σ(I): 13.3 |
| Reflection shell | Resolution: 2.28→2.4 Å / Num. unique obs: 3359 / Rrim(I) all: 0.56 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5FQD Resolution: 2.28→29.31 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.937 / Cross valid method: THROUGHOUT / ESU R: 0.214 / ESU R Free: 0.186 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 58.303 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.28→29.31 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|