 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ftf: Regulatory subunit of a cAMP-independent protein kinase A from Tr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ftf | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Regulatory subunit of a cAMP-independent protein kinase A from Trypanosoma cruzi at 1.09 A resolution | |||||||||
 Components Components | Protein kinase A regulatory subunit, putative | |||||||||
 Keywords Keywords | SIGNALING PROTEIN / protein kinase A / regulatory subunit / cell signalling | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.09151939067 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.09151939067 Å | |||||||||
 Authors Authors | Volpato Santos, Y. / Lorentzen, E. / Basquin, J. / Boshart, M. | |||||||||
| Funding support |  Brazil, Brazil,  Germany, 2items Germany, 2items
| |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2019 Title: Nucleoside analogue activators of cyclic AMP-independent protein kinase A of Trypanosoma. Authors: Bachmaier, S. / Volpato Santos, Y. / Kramer, S. / Githure, G.B. / Klockner, T. / Pepperl, J. / Baums, C. / Schenk, R. / Schwede, F. / Genieser, H.G. / Dupuy, J.W. / Forne, I. / Imhof, A. / ...Authors: Bachmaier, S. / Volpato Santos, Y. / Kramer, S. / Githure, G.B. / Klockner, T. / Pepperl, J. / Baums, C. / Schenk, R. / Schwede, F. / Genieser, H.G. / Dupuy, J.W. / Forne, I. / Imhof, A. / Basquin, J. / Lorentzen, E. / Boshart, M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ftf.cif.gz | 244.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ftf.ent.gz | 164.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6ftf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6ftf_validation.pdf.gz | 1009.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6ftf_full_validation.pdf.gz | 1013.3 KB | Display | |
| Data in XML | 6ftf_validation.xml.gz | 18.8 KB | Display | |
| Data in CIF | 6ftf_validation.cif.gz | 29.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ft/6ftfftp://data.pdbj.org/pub/pdb/validation_reports/ft/6ftf | HTTPS FTP |
-Related structure data
| Related structure data |  6floS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34476.062 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: CL Brener / Gene: Tc00.1047053506227.150 Production host:  Strain (production host): Rosetta / References: UniProt: Q4DSV5 | ||||
|---|---|---|---|---|---|
| #2: Chemical |   Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2#3: Chemical | 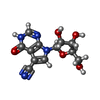  Mass: 292.247 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: C12H12N4O5 / Feature type: SUBJECT OF INVESTIGATION Mass: 292.247 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: C12H12N4O5 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 480 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 480 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.82 % Description: Most of the crystals grew in plates that poorly diffracted to 3 Angstrons , nevertheless after searching in many drops we could find more tridimensional crystals that diffracted up to 1 Angstrom |
|---|---|
| Crystal grow | Temperature: 277.15 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: Elution buffer (Size Exclusion): 50 mM Hepes pH 7.5, 50 mM NaCl, 1 mM DMSO Crystallization buffer: 17% PEG 6.000, 0.2 M calcium acetate |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06DA / Wavelength: 1 Å / Beamline: X06DA / Wavelength: 1 Å |
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: Apr 12, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.09→40.4 Å / Num. obs: 215190 / % possible obs: 81.3 % / Redundancy: 3.48 % / Biso Wilson estimate: 10.9897840654 Å2 / Net I/σ(I): 11.28 |
| Reflection shell | Resolution: 1.092→1.131 Å |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6FLO Resolution: 1.09151939067→40.4 Å / SU ML: 0.0898747422135 / Cross valid method: FREE R-VALUE / Phase error: 17.5736458365 Details: The data are >97% complete to 1.4A resolution. The low over all completeness is due to 68% completeness in the highest resolution shell (1.4-1.1A resolution
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.3294899896 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.09151939067→40.4 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -38.1723037237 Å / Origin y: 86.9790978427 Å / Origin z: 63.5542836084 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
