 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ftb: Staphylococcus aureus monofunctional glycosyltransferase in compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ftb | ||||||
|---|---|---|---|---|---|---|---|
| Title | Staphylococcus aureus monofunctional glycosyltransferase in complex with moenomycin | ||||||
 Components Components | Monofunctional glycosyltransferase | ||||||
 Keywords Keywords | TRANSFERASE / TRANSGLYCOSYLASE / PEPTIDOGLYCAN / MONOFUNCTIONAL / MOENOMYCIN / INHIBITOR / MEMBRANE / CELL SHAPE / CELL WALL BIOSYNTHESIS / GLYCOSYLTRANSFERASE / PEPTIDOGLYCAN SYNTHESIS / ANTIBIOTICS | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan glycosyltransferase / peptidoglycan glycosyltransferase activity / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / outer membrane-bounded periplasmic space / plasma membrane Similarity search - Function | ||||||
| Biological species |  Staphylococcus aureus MW2 (bacteria) Staphylococcus aureus MW2 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Punekar, A.S. / Dowson, C.J. / Roper, D.I. | ||||||
 Citation Citation | Journal: Cell Surf / Year: 2018 Title: The role of the jaw subdomain of peptidoglycan glycosyltransferases for lipid II polymerization. Authors: Punekar, A.S. / Samsudin, F. / Lloyd, A.J. / Dowson, C.G. / Scott, D.J. / Khalid, S. / Roper, D.I. #1: Journal: J Struct Biol. / Year: 2009Title: Characterization of the active site of S. aureus monofunctional glycosyltransferase (Mtg) by site-directed mutation and structural analysis of the protein complexed with moenomycin. Authors: Heaslet, H. / Shaw, B. / Mistry, A. / Miller, A.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ftb.cif.gz | 72.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ftb.ent.gz | 50.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6ftb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ft/6ftbftp://data.pdbj.org/pub/pdb/validation_reports/ft/6ftb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3hzsS S: Starting model for refinement |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 24389.998 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus MW2 (bacteria) / Gene: mgt, MW1814 / Plasmid: PPW2-SA0933(2)-N3 / Production host: References: UniProt: Q7A0I6, peptidoglycan glycosyltransferase |
|---|
-Non-polymers , 5 types, 171 molecules 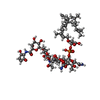




| #2: Chemical |  Mass: 1580.567 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C69H106N5O34P / Comment: antibiotic*YM Mass: 1580.567 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C69H106N5O34P / Comment: antibiotic*YM#3: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical |  Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4#5: Chemical | ChemComp-1QW / ( |  Mass: 274.396 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H30O4 Mass: 274.396 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H30O4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.35 Å3/Da / Density % sol: 63.23 % Description: HEXAGONAL PLATE CRYSTALS APPEARED WITHIN 1 WEEK. |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 4.6 Details: SA MGT E100Q (10 MG/ML) WAS MIXED WITH 1MM MOENOMYCIN AND 1MM MNCL2 AND INCUBATED ON ICE FOR ~3 HOURS. PRECIPITATED MATERIAL WAS REMOVED BY CENTRIFUGATION AT 16, 000XG FOR 5 MINUTES. THE ...Details: SA MGT E100Q (10 MG/ML) WAS MIXED WITH 1MM MOENOMYCIN AND 1MM MNCL2 AND INCUBATED ON ICE FOR ~3 HOURS. PRECIPITATED MATERIAL WAS REMOVED BY CENTRIFUGATION AT 16, 000XG FOR 5 MINUTES. THE MOENOMYCIN COMPLEX WAS CRYSTALLIZED BY HANGING DROP VAPOR DIFFUSION, MIXING THE PROTEIN 1:1 WITH A RESERVOIR SOLUTION CONTAINING 0.1M NA ACETATE PH 4.6, 0.2M NACL, 30% MPD AT 295 K. PH range: 4.6 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E DW / Wavelength: 1.54 Å |
| Detector | Type: RIGAKU SATURN 944 / Detector: CCD / Date: Sep 12, 2007 / Details: RIGAKU VARIMAX HF OPTICS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→23.12 Å / Num. obs: 45531 / % possible obs: 99.9 % / Observed criterion σ(I): 2 / Redundancy: 3.9 % / Biso Wilson estimate: 37.46 Å2 / Rsym value: 0.173 / Net I/σ(I): 13.6 |
| Reflection shell | Resolution: 2.1→2.2 Å / Redundancy: 3.8 % / Mean I/σ(I) obs: 3.2 / Rsym value: 0.381 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3HZS Resolution: 2.1→23.12 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.924 / Rfactor Rfree error: 0 / SU R Cruickshank DPI: 0.184 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.193 / SU Rfree Blow DPI: 0.168 / SU Rfree Cruickshank DPI: 0.165
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 42.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.26 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→23.12 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.21 Å / Rfactor Rfree error: 0 / Total num. of bins used: 10
|
