 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3dwk: Identification of Dynamic Structural Motifs Involved in Peptidogl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3dwk | ||||||
|---|---|---|---|---|---|---|---|
| Title | Identification of Dynamic Structural Motifs Involved in Peptidoglycan Glycosyltransfer | ||||||
 Components Components | Penicillin-binding protein 2 | ||||||
 Keywords Keywords | TRANSFERASE / lysozyme-fold transpeptidase fold pi-helix / Cell shape / Cell wall biogenesis/degradation / Membrane / Peptidoglycan synthesis | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan glycosyltransferase / peptidoglycan glycosyltransferase activity / serine-type D-Ala-D-Ala carboxypeptidase / serine-type D-Ala-D-Ala carboxypeptidase activity / penicillin binding / membrane => GO:0016020 / peptidoglycan biosynthetic process / outer membrane-bounded periplasmic space / proteolysis Similarity search - Function | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 3.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 3.1 Å | ||||||
 Authors Authors | Lovering, A.L. / De Castro, L. / Strynadka, N.C.J. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2008 Title: Identification of dynamic structural motifs involved in peptidoglycan glycosyltransfer. Authors: Lovering, A.L. / De Castro, L. / Strynadka, N.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3dwk.cif.gz | 426.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3dwk.ent.gz | 355.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3dwk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dw/3dwkftp://data.pdbj.org/pub/pdb/validation_reports/dw/3dwk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2olvS S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 69626.914 Da / Num. of mol.: 4 / Fragment: PBP2 (UNP residues 68-692) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria) / Gene: pbp2, SACOL1490 / Plasmid: pET41 / Production host: #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 46 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 46 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-LDA / | 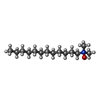  Mass: 229.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM Mass: 229.402 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H31NO / Comment: LDAO, detergent*YM |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.19 Å3/Da / Density % sol: 61.44 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 100mM Hepes pH 7.5 2M Ammonium Sulphate 0.28mM LDAO, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.2.2 / Wavelength: 0.96649 Å / Beamline: 8.2.2 / Wavelength: 0.96649 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 1, 2006 |
| Radiation | Monochromator: Double crystal, Si / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.96649 Å / Relative weight: 1 |
| Reflection | Resolution: 3.1→41.88 Å / Num. all: 63100 / Num. obs: 60450 / % possible obs: 95.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.9 % / Rsym value: 0.119 / Net I/σ(I): 8.2 |
| Reflection shell | Resolution: 3.1→3.27 Å / Redundancy: 2.6 % / Rmerge(I) obs: 0.517 / Mean I/σ(I) obs: 1.9 / % possible all: 92.4 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 2OLV Resolution: 3.1→41.88 Å / Occupancy max: 1 / Occupancy min: 1 / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 317.12 Å2 / Biso mean: 72.405 Å2 / Biso min: 8.12 Å2 | ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.1→41.88 Å
|
