 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6dnl: Crystal Structure of Neisseria meningitidis DsbD c-terminal domai... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6dnl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Neisseria meningitidis DsbD c-terminal domain in the reduced form | ||||||
 Components Components | Thiol:disulfide interchange protein DsbD | ||||||
 Keywords Keywords | OXIDOREDUCTASE / disulphide reductase / Dsb proteins | ||||||
| Function / homology |  Function and homology information Function and homology informationprotein-disulfide reductase / protein-disulfide reductase [NAD(P)H] activity / cytochrome complex assembly / protein-disulfide reductase activity / membrane => GO:0016020 / cell redox homeostasis / electron transfer activity / plasma membrane Similarity search - Function | ||||||
| Biological species |  Neisseria meningitidis (bacteria) Neisseria meningitidis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.7 Å | ||||||
 Authors Authors | Smith, R.P. / Heras, B. / Paxman, J.J. | ||||||
 Citation Citation | Journal: J. Biol. Chem. / Year: 2018 Title: Structural and biochemical insights into the disulfide reductase mechanism of DsbD, an essential enzyme for neisserial pathogens. Authors: Smith, R.P. / Mohanty, B. / Mowlaboccus, S. / Paxman, J.J. / Williams, M.L. / Headey, S.J. / Wang, G. / Subedi, P. / Doak, B.C. / Kahler, C.M. / Scanlon, M.J. / Heras, B. #1: Journal: Acta Crystallogr F Struct Biol Commun / Year: 2018 Title: Production, biophysical characterization and initial crystallization studies of the N- and C-terminal domains of DsbD, an essential enzyme in Neisseria meningitidis. Authors: Smith, R.P. / Whitten, A.E. / Paxman, J.J. / Kahler, C.M. / Scanlon, M.J. / Heras, B. | ||||||
| History |
|
- Structure visualization
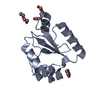
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6dnl.cif.gz | 66.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6dnl.ent.gz | 47.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6dnl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dn/6dnlftp://data.pdbj.org/pub/pdb/validation_reports/dn/6dnl | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13100.884 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Neisseria meningitidis (bacteria) / Gene: dipZ, dsbD, A6L27_07425, ERS514851_00843 / Plasmid: pMCSG7Production host: References: UniProt: A0A0Y6T358, UniProt: Q9JYM0*PLUS, protein-disulfide reductase | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn#3: Chemical |   Mass: 59.044 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H3O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 94 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 94 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 40.94 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 7.5 Details: 0.2 M zinc acetate dehydrate, 0.1 M sodium cacodylate trihydrate pH 7.5 and 18% w/v PEG 8000 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron  / Beamline: MX2 / Wavelength: 0.9537 Å / Beamline: MX2 / Wavelength: 0.9537 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Sep 30, 2016 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9537 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.7→44.965 Å / Num. obs: 12150 / % possible obs: 99.8 % / Redundancy: 3.8 % / Biso Wilson estimate: 18.71 Å2 / Rpim(I) all: 0.059 / Rrim(I) all: 0.116 / Rsym value: 0.099 / Net I/av σ(I): 5 / Net I/σ(I): 7.5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.7→34.298 Å / SU ML: 0.2 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 22.45
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 148.14 Å2 / Biso mean: 28.3486 Å2 / Biso min: 9.54 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.7→34.298 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 5
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
