Journal: J Biol Chem / Year: 2018 Title: Common fibrillar spines of amyloid-β and human islet amyloid polypeptide revealed by microelectron diffraction and structure-based inhibitors. Authors: Pascal Krotee / Sarah L Griner / Michael R Sawaya / Duilio Cascio / Jose A Rodriguez / Dan Shi / Stephan Philipp / Kevin Murray / Lorena Saelices / Ji Lee / Paul Seidler / Charles G Glabe / ...Authors: Pascal Krotee / Sarah L Griner / Michael R Sawaya / Duilio Cascio / Jose A Rodriguez / Dan Shi / Stephan Philipp / Kevin Murray / Lorena Saelices / Ji Lee / Paul Seidler / Charles G Glabe / Lin Jiang / Tamir Gonen / David S Eisenberg / Abstract: Amyloid-β (Aβ) and human islet amyloid polypeptide (hIAPP) aggregate to form amyloid fibrils that deposit in tissues and are associated with Alzheimer's disease (AD) and type II diabetes (T2D), ...Amyloid-β (Aβ) and human islet amyloid polypeptide (hIAPP) aggregate to form amyloid fibrils that deposit in tissues and are associated with Alzheimer's disease (AD) and type II diabetes (T2D), respectively. Individuals with T2D have an increased risk of developing AD, and conversely, AD patients have an increased risk of developing T2D. Evidence suggests that this link between AD and T2D might originate from a structural similarity between aggregates of Aβ and hIAPP. Using the cryoEM method microelectron diffraction, we determined the atomic structures of 11-residue segments from both Aβ and hIAPP, termed Aβ(24-34) WT and hIAPP(19-29) S20G, with 64% sequence similarity. We observed a high degree of structural similarity between their backbone atoms (0.96-Å root mean square deviation). Moreover, fibrils of these segments induced amyloid formation through self- and cross-seeding. Furthermore, inhibitors designed for one segment showed cross-efficacy for full-length Aβ and hIAPP and reduced cytotoxicity of both proteins, although by apparently blocking different cytotoxic mechanisms. The similarity of the atomic structures of Aβ(24-34) WT and hIAPP(19-29) S20G offers a molecular model for cross-seeding between Aβ and hIAPP.
∠α: 90 ° / ∠β: 100.017 ° / ∠γ: 90 ° / A: 18.78 Å / B: 4.73 Å / C: 33.47 Å / Space group name: P21 / Space group num: 4
CTF correction
Type: NONE
3D reconstruction
Resolution method: DIFFRACTION PATTERN/LAYERLINES / Symmetry type: 3D CRYSTAL
Atomic model building
Protocol: OTHER / Space: RECIPROCAL / Target criteria: Maximum likelihood
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.42→32.96 Å / Cor.coef. Fo:Fc: 0.933 / Cor.coef. Fo:Fc free: 0.881 / SU B: 2.871 / SU ML: 0.1 / SU R Cruickshank DPI: 0.1152 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.115 / ESU R Free: 0.122 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2924
113
10 %
RANDOM
Rwork
0.2335
-
-
-
obs
0.2389
1014
85.51 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) MOLECULAR REPLACEMENT /
MOLECULAR REPLACEMENT /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads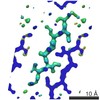







 PDBj
PDBj













 Assembly
Assembly

 Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation
 Processing
Processing