National Institutes of Health/National Institute on Aging (NIH/NIA)
R01 AG029430
United States
Howard Hughes Medical Institute (HHMI)
United States
Citation
Journal: Elife / Year: 2017 Title: Atomic structures of fibrillar segments of hIAPP suggest tightly mated β-sheets are important for cytotoxicity. Authors: Pascal Krotee / Jose A Rodriguez / Michael R Sawaya / Duilio Cascio / Francis E Reyes / Dan Shi / Johan Hattne / Brent L Nannenga / Marie E Oskarsson / Stephan Philipp / Sarah Griner / Lin ...Authors: Pascal Krotee / Jose A Rodriguez / Michael R Sawaya / Duilio Cascio / Francis E Reyes / Dan Shi / Johan Hattne / Brent L Nannenga / Marie E Oskarsson / Stephan Philipp / Sarah Griner / Lin Jiang / Charles G Glabe / Gunilla T Westermark / Tamir Gonen / David S Eisenberg / Abstract: hIAPP fibrils are associated with Type-II Diabetes, but the link of hIAPP structure to islet cell death remains elusive. Here we observe that hIAPP fibrils are cytotoxic to cultured pancreatic β- ...hIAPP fibrils are associated with Type-II Diabetes, but the link of hIAPP structure to islet cell death remains elusive. Here we observe that hIAPP fibrils are cytotoxic to cultured pancreatic β-cells, leading us to determine the structure and cytotoxicity of protein segments composing the amyloid spine of hIAPP. Using the cryoEM method MicroED, we discover that one segment, 19-29 S20G, forms pairs of β-sheets mated by a dry interface that share structural features with and are similarly cytotoxic to full-length hIAPP fibrils. In contrast, a second segment, 15-25 WT, forms non-toxic labile β-sheets. These segments possess different structures and cytotoxic effects, however, both can seed full-length hIAPP, and cause hIAPP to take on the cytotoxic and structural features of that segment. These results suggest that protein segment structures represent polymorphs of their parent protein and that segment 19-29 S20G may serve as a model for the toxic spine of hIAPP.
The biological unit is an extended pair of beta sheets comprising peptides at positions X,Y,Z and 1/2+X,-1/2-Y,-Z repeated ad infinitum along the a crystal axis.
Instrument: 1.5 mL Eppendorf tube / Atmosphere: Air, sealed chamber. Details: 1 mM lyophilized peptide in PBS with 1% DMSO at room temperature under quiescent conditions. Crystals grew in a few hours and reached full size within 15 hours. Temperature: 298 K / Time: 2 DAY
Buffer solution
pH: 7.4
Buffer component
ID
Conc.
Name
Buffer-ID
1
phosphate-buffered saline
1
2
1 %
DMSO
1
Specimen
Conc.: 1.066 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES
Temperature: 295 K / Method: batch mode / pH: 7.4 Details: 1 mmol SGNNFGAILSS in 1 L phosphate-buffered saline with 1% DMSO incubated under quiescent conditions overnight
-
Data collection
Microscopy
Model: FEI TECNAI 20
Electron gun
Electron source: FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM
Electron lens
Mode: DIFFRACTION / Alignment procedure: BASIC
Specimen holder
Cryogen: NITROGEN Specimen holder model: GATAN 626 SINGLE TILT LIQUID NITROGEN CRYO TRANSFER HOLDER Temperature (max): 100 K / Temperature (min): 100 K
Image recording
Average exposure time: 2 sec. / Electron dose: 0.01 e/Å2 / Film or detector model: TVIPS TEMCAM-F416 (4k x 4k) / Num. of diffraction images: 879 / Num. of grids imaged: 2 Details: The detector was operated in rolling shutter mode with 2x2 pixel binning.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) MOLECULAR REPLACEMENT /
MOLECULAR REPLACEMENT /  Authors
Authors United States, 2items
United States, 2items  Citation
Citation

 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads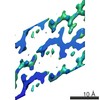










 PDBj
PDBj



 Assembly
Assembly

 Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing