 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5ung: XFEL structure of human angiotensin II type 2 receptor (Orthorhom... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5ung | ||||||
|---|---|---|---|---|---|---|---|
| Title | XFEL structure of human angiotensin II type 2 receptor (Orthorhombic form) in complex with compound 1 (N-benzyl-N-(2-ethyl-4-oxo-3-{[2'-(2H-tetrazol-5-yl)[1,1'-biphenyl]-4-yl] methyl}-3,4-dihydroquinazolin-6-yl)thiophene-2-carboxamide) | ||||||
 Components Components | Chimera protein of Type-2 angiotensin II receptor and Soluble cytochrome b562 | ||||||
 Keywords Keywords | SIGNALING PROTEIN / human Angiotensin II receptor complex / GPCR signaling / GPCR / BRIL / membrane protein / LCP / XFEL / blood pressure regulation / orthorhombic crystal / compound 1 (cpd 1) | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of metanephros size / regulation of systemic arterial blood pressure by circulatory renin-angiotensin / brain renin-angiotensin system / angiotensin type II receptor activity / angiotensin-mediated vasodilation involved in regulation of systemic arterial blood pressure / negative regulation of neurotrophin TRK receptor signaling pathway / positive regulation of metanephric glomerulus development / receptor antagonist activity / G protein-coupled receptor signaling pathway coupled to cGMP nucleotide second messenger / positive regulation of branching involved in ureteric bud morphogenesis ...regulation of metanephros size / regulation of systemic arterial blood pressure by circulatory renin-angiotensin / brain renin-angiotensin system / angiotensin type II receptor activity / angiotensin-mediated vasodilation involved in regulation of systemic arterial blood pressure / negative regulation of neurotrophin TRK receptor signaling pathway / positive regulation of metanephric glomerulus development / receptor antagonist activity / G protein-coupled receptor signaling pathway coupled to cGMP nucleotide second messenger / positive regulation of branching involved in ureteric bud morphogenesis / positive regulation of extrinsic apoptotic signaling pathway / negative regulation of heart rate / exploration behavior / negative regulation of blood vessel endothelial cell migration / blood vessel remodeling / nitric oxide-cGMP-mediated signaling / Peptide ligand-binding receptors / electron transport chain / negative regulation of cell growth / brain development / regulation of blood pressure / vasodilation / neuron apoptotic process / G alpha (i) signalling events / electron transfer activity / periplasmic space / cell surface receptor signaling pathway / iron ion binding / G protein-coupled receptor signaling pathway / inflammatory response / heme binding / positive regulation of DNA-templated transcription / plasma membrane Similarity search - Function | ||||||
| Biological species |   Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / FREE ELECTRON LASER / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / FREE ELECTRON LASER / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Zhang, H. / Han, G.W. / Batyuk, A. / Ishchenko, A. / White, K.L. / Patel, N. / Sadybekov, A. / Zamlynny, B. / Rudd, M.T. / Hollenstein, K. ...Zhang, H. / Han, G.W. / Batyuk, A. / Ishchenko, A. / White, K.L. / Patel, N. / Sadybekov, A. / Zamlynny, B. / Rudd, M.T. / Hollenstein, K. / Tolstikova, A. / White, T.A. / Hunter, M.S. / Weierstall, U. / Liu, W. / Babaoglu, K. / Moore, E.L. / Katz, R.D. / Shipman, J.M. / Garcia-Calvo, M. / Sharma, S. / Sheth, P. / Soisson, S.M. / Stevens, R.C. / Katritch, V. / Cherezov, V. | ||||||
 Citation Citation | Journal: Nature / Year: 2017 Title: Structural basis for selectivity and diversity in angiotensin II receptors. Authors: Zhang, H. / Han, G.W. / Batyuk, A. / Ishchenko, A. / White, K.L. / Patel, N. / Sadybekov, A. / Zamlynny, B. / Rudd, M.T. / Hollenstein, K. / Tolstikova, A. / White, T.A. / Hunter, M.S. / ...Authors: Zhang, H. / Han, G.W. / Batyuk, A. / Ishchenko, A. / White, K.L. / Patel, N. / Sadybekov, A. / Zamlynny, B. / Rudd, M.T. / Hollenstein, K. / Tolstikova, A. / White, T.A. / Hunter, M.S. / Weierstall, U. / Liu, W. / Babaoglu, K. / Moore, E.L. / Katz, R.D. / Shipman, J.M. / Garcia-Calvo, M. / Sharma, S. / Sheth, P. / Soisson, S.M. / Stevens, R.C. / Katritch, V. / Cherezov, V. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5ung.cif.gz | 181.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5ung.ent.gz | 141.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5ung.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/un/5ungftp://data.pdbj.org/pub/pdb/validation_reports/un/5ung | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5unfC  5unhC  4yayS  4zudS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | AUTHORS STATE THAT THE BIOLOGICAL UNIT IS UNKNOWN |
-Components
| #1: Protein | Mass: 46506.535 Da / Num. of mol.: 1 Fragment: UNP P0ABE7 residues 23-128 and UNP P50052 35-335 linked via LINKER resdiues GSGS Mutation: M1007W, H1102I, R1106L Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human)Gene: cybC, AGTR2 / Production host:   Spodoptera frugiperda (fall armyworm) / Strain (production host): sf9 / References: UniProt: P0ABE7, UniProt: P50052 Spodoptera frugiperda (fall armyworm) / Strain (production host): sf9 / References: UniProt: P0ABE7, UniProt: P50052 |
|---|---|
| #2: Chemical | ChemComp-8ES /   Mass: 623.726 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C36H29N7O2S Mass: 623.726 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C36H29N7O2S |
| #3: Chemical | ChemComp-OLC / (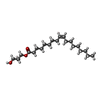  Mass: 356.540 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H40O4 Mass: 356.540 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H40O4 |
| #4: Chemical | ChemComp-OLA / 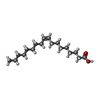  Mass: 282.461 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H34O2 Mass: 282.461 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H34O2 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.78 Å3/Da / Density % sol: 55.75 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: lipidic cubic phase / pH: 8 Details: 100 mM Tris-HCl, pH 8.0, 25 mM potassium formate, 25% (v/v) PEG400, and 0.3% (v/v) (+/-)-2-Methyl-2,4-pentanediol |
-Data collection
| Diffraction | Mean temperature: 294 K / Serial crystal experiment: Y |
|---|---|
| Diffraction source | Source: FREE ELECTRON LASER / Site: SLAC LCLS  / Beamline: CXI / Wavelength: 1.3 Å / Beamline: CXI / Wavelength: 1.3 Å |
| Detector | Type: CS-PAD CXI-1 / Detector: PIXEL / Date: Dec 9, 2015 / Details: K-B mirrors |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.3 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→28.96 Å / Num. obs: 13330 / % possible obs: 100 % / Redundancy: 85.5 % / Biso Wilson estimate: 80.91 Å2 / Rmerge(I) obs: 0.148 / Net I/σ(I): 4.9 |
| Reflection shell | Resolution: 2.8→2.9 Å / Redundancy: 27.2 % / Rmerge(I) obs: 1.24 / Mean I/σ(I) obs: 1 / % possible all: 100 |
| Serial crystallography measurement | Photons per pulse: 1011 Tphotons/pulse / Pulse duration: 40 fsec. / Pulse energy: 1.8 µJ / XFEL pulse repetition rate: 120 Hz |
| Serial crystallography sample delivery | Method: injection |
| Serial crystallography data reduction | Crystal hits: 175241 / Frames indexed: 15804 / Frames total: 2701530 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4YAY, 4ZUD Resolution: 2.8→28.96 Å / Cor.coef. Fo:Fc: 0.912 / Cor.coef. Fo:Fc free: 0.881 / SU R Cruickshank DPI: 2.685 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 2.894 / SU Rfree Blow DPI: 0.357 / SU Rfree Cruickshank DPI: 0.363
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 82.84 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.49 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.8→28.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→3.02 Å / Rfactor Rfree error: 0 / Total num. of bins used: 7
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
