Entry Database : PDB / ID : 4zudTitle Crystal Structure of Human Angiotensin Receptor in Complex with Inverse Agonist Olmesartan at 2.8A resolution. Chimera protein of Soluble cytochrome b562 and Type-1 angiotensin II receptor Keywords / / / / / / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Escherichia coli (E. coli)Homo sapiens (human)Method / / / Resolution : 2.8 Å Authors Zhang, H. / Unal, H. / Desnoyer, R. / Han, G.W. / Patel, N. / Katritch, V. / Karnik, S.S. / Cherezov, V. / Stevens, R.C. / GPCR Network (GPCR) Funding support Organization Grant number Country National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) U54 GM094618
Journal : J.Biol.Chem. / Year : 2015Title : Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor.Authors : Zhang, H. / Unal, H. / Desnoyer, R. / Han, G.W. / Patel, N. / Katritch, V. / Karnik, S.S. / Cherezov, V. / Stevens, R.C. History Deposition May 15, 2015 Deposition site / Processing site Revision 1.0 Oct 7, 2015 Provider / Type Revision 1.1 Oct 14, 2015 Group Revision 1.2 Dec 16, 2015 Group Revision 1.3 Sep 20, 2017 Group / Database references / Derived calculationsCategory / pdbx_audit_support / pdbx_struct_oper_listItem / _pdbx_audit_support.funding_organization / _pdbx_struct_oper_list.symmetry_operationRevision 1.4 Dec 25, 2019 Group / Category / Item Revision 1.5 Sep 27, 2023 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accessionRevision 1.6 Nov 6, 2024 Group / Category / pdbx_modification_feature
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj















 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm) / Strain (production host): sf9 / References: UniProt: P0ABE7, UniProt: P30556
Spodoptera frugiperda (fall armyworm) / Strain (production host): sf9 / References: UniProt: P0ABE7, UniProt: P30556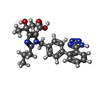
 Mass: 446.502 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H26N6O3
Mass: 446.502 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H26N6O3 Sample preparation
Sample preparation Processing
Processing