登録情報 データベース : PDB / ID : 5nf0タイトル Discovery, crystal structures and atomic force microscopy study of thioether ligated D,L-cyclic antimicrobial peptides against multidrug resistant Pseudomonas aeruginosa CYD-TRP-TRD-LYS-LYD-LYS-LYD-LYS-TRD-TRP-CYD-GLY Fragment of ligand Fucose-binding lectin II (PA-IIL) キーワード / / / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / / / 生物種 Pseudomonas aeruginosa (緑膿菌)synthetic construct (人工物) 手法 / / / 解像度 : 1.271 Å データ登録者 Reymond, J.-L. / Darbre, T. / Stocker, A. / Hong, W. / van Delden, C. / Koehler, T. / Luscher, A. / Visini, R. / Fu, Y. / Di Bonaventura, I. / He, R. ジャーナル : Chem Sci / 年 : 2017タイトル : Design, crystal structure and atomic force microscopy study of thioether ligated d,l-cyclic antimicrobial peptides against multidrug resistant Pseudomonas aeruginosa.著者 : He, R. / Di Bonaventura, I. / Visini, R. / Gan, B.H. / Fu, Y. / Probst, D. / Luscher, A. / Kohler, T. / van Delden, C. / Stocker, A. / Hong, W. / Darbre, T. / Reymond, J.L. 履歴 登録 2017年3月13日 登録サイト / 処理サイト 改定 1.0 2017年9月13日 Provider / タイプ 改定 1.1 2017年12月6日 Group / Derived calculationsカテゴリ / citation_author / pdbx_struct_assembly_genItem _citation.journal_id_ISSN / _citation.journal_volume ... _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation_author.name / _pdbx_struct_assembly_gen.asym_id_list 改定 1.2 2020年7月29日 Group / Derived calculations / Refinement descriptionカテゴリ chem_comp / pdbx_struct_conn_angle ... chem_comp / pdbx_struct_conn_angle / refine_hist / struct_conn / struct_conn_type / struct_site / struct_site_gen Item _chem_comp.type / _pdbx_struct_conn_angle.ptnr1_auth_asym_id ... _chem_comp.type / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _refine_hist.d_res_low / _struct_conn.conn_type_id / _struct_conn.id / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn_type.id 解説 / Provider / タイプ 改定 1.3 2024年1月17日 Group / Database references / Refinement descriptionカテゴリ chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession改定 2.0 2025年10月1日 Group Advisory / Atomic model ... Advisory / Atomic model / Data collection / Derived calculations / Other / Structure summary カテゴリ atom_site / pdbx_database_status ... atom_site / pdbx_database_status / pdbx_entry_details / pdbx_modification_feature / pdbx_nonpoly_scheme / pdbx_struct_conn_angle / pdbx_struct_sheet_hbond / pdbx_unobs_or_zero_occ_atoms / pdbx_validate_close_contact / struct_asym / struct_conn / struct_sheet / struct_sheet_order / struct_sheet_range Item _atom_site.B_iso_or_equiv / _atom_site.Cartn_x ... _atom_site.B_iso_or_equiv / _atom_site.Cartn_x / _atom_site.Cartn_y / _atom_site.Cartn_z / _atom_site.auth_asym_id / _atom_site.auth_atom_id / _atom_site.auth_comp_id / _atom_site.auth_seq_id / _atom_site.label_asym_id / _atom_site.label_atom_id / _atom_site.label_comp_id / _atom_site.label_entity_id / _atom_site.type_symbol / _pdbx_database_status.pdb_format_compatible / _pdbx_nonpoly_scheme.asym_id / _pdbx_nonpoly_scheme.auth_mon_id / _pdbx_nonpoly_scheme.auth_seq_num / _pdbx_nonpoly_scheme.entity_id / _pdbx_nonpoly_scheme.mon_id / _pdbx_nonpoly_scheme.ndb_seq_num / _pdbx_nonpoly_scheme.pdb_mon_id / _pdbx_nonpoly_scheme.pdb_seq_num / _pdbx_nonpoly_scheme.pdb_strand_id / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_auth_asym_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_asym.entity_id
すべて表示 表示を減らす
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Pseudomonas aeruginosa (緑膿菌)
Pseudomonas aeruginosa (緑膿菌) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード










 PDBj
PDBj 集合体
集合体

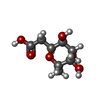
 タイプ: D-saccharide / 分子量: 206.193 Da / 分子数: 4 / 由来タイプ: 組換発現 / 式: C8H14O6
タイプ: D-saccharide / 分子量: 206.193 Da / 分子数: 4 / 由来タイプ: 組換発現 / 式: C8H14O6

 分子量: 40.078 Da / 分子数: 8 / 由来タイプ: 合成 / 式: Ca
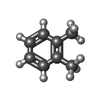
分子量: 40.078 Da / 分子数: 8 / 由来タイプ: 合成 / 式: Ca 分子量: 106.165 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C8H10
分子量: 106.165 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C8H10 試料調製
試料調製 / ビームライン: X06DA / 波長: 1.00003 Å
/ ビームライン: X06DA / 波長: 1.00003 Å 解析
解析