 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5mfq: Crystal structure of the GluK1 ligand-binding domain in complex w... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5mfq | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the GluK1 ligand-binding domain in complex with kainate and BPAM-344 at 1.90 A resolution | |||||||||
 Components Components | Glutamate receptor ionotropic, kainate 1,Glutamate receptor ionotropic, kainate 1 | |||||||||
 Keywords Keywords | MEMBRANE PROTEIN / ionotropic glutamate receptor / GluK1 ligand-binding domain / positive allosteric modulator | |||||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of synaptic transmission, GABAergic / gamma-aminobutyric acid secretion / L-glutamate transmembrane transporter activity / amino acid transmembrane transport / positive regulation of gamma-aminobutyric acid secretion / Activation of Na-permeable kainate receptors / kainate selective glutamate receptor complex / Activation of Ca-permeable Kainate Receptor / negative regulation of synaptic transmission, glutamatergic / regulation of short-term neuronal synaptic plasticity ...negative regulation of synaptic transmission, GABAergic / gamma-aminobutyric acid secretion / L-glutamate transmembrane transporter activity / amino acid transmembrane transport / positive regulation of gamma-aminobutyric acid secretion / Activation of Na-permeable kainate receptors / kainate selective glutamate receptor complex / Activation of Ca-permeable Kainate Receptor / negative regulation of synaptic transmission, glutamatergic / regulation of short-term neuronal synaptic plasticity / inhibitory postsynaptic potential / synaptic transmission, GABAergic / glutamate binding / adult behavior / behavioral response to pain / modulation of excitatory postsynaptic potential / extracellularly glutamate-gated ion channel activity / membrane depolarization / kainate selective glutamate receptor activity / ionotropic glutamate receptor complex / sodium ion transmembrane transport / glutamate-gated receptor activity / glutamate-gated calcium ion channel activity / ligand-gated monoatomic ion channel activity involved in regulation of presynaptic membrane potential / potassium ion transmembrane transport / presynaptic modulation of chemical synaptic transmission / ionotropic glutamate receptor signaling pathway / regulation of membrane potential / SNARE binding / excitatory postsynaptic potential / synaptic transmission, glutamatergic / establishment of localization in cell / positive regulation of synaptic transmission, GABAergic / transmitter-gated monoatomic ion channel activity involved in regulation of postsynaptic membrane potential / postsynaptic density membrane / modulation of chemical synaptic transmission / regulation of synaptic plasticity / terminal bouton / presynaptic membrane / nervous system development / scaffold protein binding / chemical synaptic transmission / postsynaptic membrane / postsynaptic density / receptor complex / neuronal cell body / glutamatergic synapse / dendrite / synapse / identical protein binding / membrane / plasma membrane Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.9 Å | |||||||||
 Authors Authors | Larsen, A.P. / Frydenvang, K. / Kastrup, J.S. | |||||||||
 Citation Citation | Journal: Mol. Pharmacol. / Year: 2017 Title: Identification and Structure-Function Study of Positive Allosteric Modulators of Kainate Receptors. Authors: Larsen, A.P. / Fievre, S. / Frydenvang, K. / Francotte, P. / Pirotte, B. / Kastrup, J.S. / Mulle, C. #1: Journal: FEBS Lett. / Year: 2005Title: Crystal structure of the kainate receptor GluR5 ligand-binding core in complex with (S)-glutamate. Authors: Naur, P. / Vestergaard, B. / Skov, L.K. / Egebjerg, J. / Gajhede, M. / Kastrup, J.S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5mfq.cif.gz | 230.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5mfq.ent.gz | 182.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5mfq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5mfq_validation.pdf.gz | 486.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5mfq_full_validation.pdf.gz | 489.8 KB | Display | |
| Data in XML | 5mfq_validation.xml.gz | 27.1 KB | Display | |
| Data in CIF | 5mfq_validation.cif.gz | 40.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mf/5mfqftp://data.pdbj.org/pub/pdb/validation_reports/mf/5mfq | HTTPS FTP |
-Related structure data
| Related structure data |  5mfvC  5mfwC  4e0xS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 29108.453 Da / Num. of mol.: 2 Fragment: UNP residues 445-559,UNP residues 682-820,UNP residues 445-559,UNP residues 682-820 Source method: isolated from a genetically manipulated source Details: THE PROTEIN CRYSTALLIZED IS THE EXTRACELLULAR LIGAND BINDING DOMAIN OF GLUK1. TRANSMEMBRANE REGIONS WERE GENETICALLY REMOVED AND REPLACED WITH A GLY-THR LINKER (RESIDUES 545 AND 546 OF THE ...Details: THE PROTEIN CRYSTALLIZED IS THE EXTRACELLULAR LIGAND BINDING DOMAIN OF GLUK1. TRANSMEMBRANE REGIONS WERE GENETICALLY REMOVED AND REPLACED WITH A GLY-THR LINKER (RESIDUES 545 AND 546 OF THE STRUCTURE). THEREFORE, THE SEQUENCE MATCHES DISCONTINUOUSLY WITH THE REFERENCE DATABASE (430-544, 667-805). RESIDUE 429 IS A REMNANT FROM CLONING.,THE PROTEIN CRYSTALLIZED IS THE EXTRACELLULAR LIGAND BINDING DOMAIN OF GLUK1. TRANSMEMBRANE REGIONS WERE GENETICALLY REMOVED AND REPLACED WITH A GLY-THR LINKER (RESIDUES 545 AND 546 OF THE STRUCTURE). THEREFORE, THE SEQUENCE MATCHES DISCONTINUOUSLY WITH THE REFERENCE DATABASE (430-544, 667-805). RESIDUE 429 IS A REMNANT FROM CLONING. Source: (gene. exp.)  |
|---|
-Non-polymers , 6 types, 592 molecules 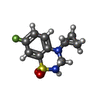





| #2: Chemical |  Mass: 242.270 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H11FN2O2S Mass: 242.270 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H11FN2O2S#3: Chemical |  Mass: 213.230 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15NO4 / Comment: neurotransmitter, agonist*YM Mass: 213.230 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15NO4 / Comment: neurotransmitter, agonist*YM#4: Chemical | ChemComp-CL / |  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#5: Chemical |  Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#6: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 582 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|---|
| Sequence details | THE SEQUENCE DATABASE IS P22756-2, ISOFORM GLUR5-2.THE PROTEIN CRYSTALLIZED IS THE EXTRACELLULAR ...THE SEQUENCE DATABASE IS P22756-2, ISOFORM GLUR5-2.THE PROTEIN CRYSTALLIZ |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.56 Å3/Da / Density % sol: 52.03 % |
|---|---|
| Crystal grow | Temperature: 279 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: 15.2 % PEG4000, 0.3 M lithium-sulfate, 0.1 M sodium-acetate pH 5.5 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II  / Beamline: I911-3 / Wavelength: 0.97916 Å / Beamline: I911-3 / Wavelength: 0.97916 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Feb 28, 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97916 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.9→29.429 Å / Num. obs: 48600 / % possible obs: 100 % / Redundancy: 8.1 % / Biso Wilson estimate: 18.22 Å2 / Rsym value: 0.072 / Net I/av σ(I): 8.04 / Net I/σ(I): 18.6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Rfactor: 40.45 / Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 4E0X Resolution: 1.9→29.429 Å / SU ML: 0.15 / Cross valid method: FREE R-VALUE / σ(F): 0 / Phase error: 17.72
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 85.94 Å2 / Biso mean: 23.11 Å2 / Biso min: 5.17 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.9→29.429 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 18
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
