| Entry | Database: PDB / ID: 5i0b
|
|---|
| Title | Structure of PAK4 |
|---|
 Components Components | Serine/threonine-protein kinase PAK 4 |
|---|
 Keywords Keywords | TRANSFERASE/TRANSFERASE INIHIBITOR / kinase / TRANSFERASE-TRANSFERASE INIHIBITOR complex |
|---|
| Function / homology |  Function and homology information Function and homology information
dendritic spine development / cadherin binding involved in cell-cell adhesion / Activation of RAC1 / RHOV GTPase cycle / RHOJ GTPase cycle / RHOQ GTPase cycle / RHOU GTPase cycle / regulation of MAPK cascade / CDC42 GTPase cycle / RHOH GTPase cycle ...dendritic spine development / cadherin binding involved in cell-cell adhesion / Activation of RAC1 / RHOV GTPase cycle / RHOJ GTPase cycle / RHOQ GTPase cycle / RHOU GTPase cycle / regulation of MAPK cascade / CDC42 GTPase cycle / RHOH GTPase cycle / RHOG GTPase cycle / RAC2 GTPase cycle / RAC3 GTPase cycle / negative regulation of endothelial cell apoptotic process / RAC1 GTPase cycle / cytoskeleton organization / cellular response to starvation / adherens junction / regulation of cell growth / positive regulation of angiogenesis / cell migration / non-specific serine/threonine protein kinase / protein kinase activity / intracellular signal transduction / protein serine kinase activity / focal adhesion / protein serine/threonine kinase activity / apoptotic process / Golgi apparatus / signal transduction / ATP binding / cytoplasm / cytosolSimilarity search - Function p21 activated kinase binding domain / : / CRIB domain superfamily / P21-Rho-binding domain / CRIB domain profile. / P21-Rho-binding domain / CRIB domain / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 ...p21 activated kinase binding domain / : / CRIB domain superfamily / P21-Rho-binding domain / CRIB domain profile. / P21-Rho-binding domain / CRIB domain / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Protein kinase domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.09 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.09 Å |
|---|
 Authors Authors | Park, S.Y. |
|---|
| Funding support |  Korea, Republic Of, 1items Korea, Republic Of, 1items | Organization | Grant number | Country |
|---|
| National Research Foundation of Korea | 2013R1A1A2005276 | Korea, Republic Of |
|
|---|
 Citation Citation | Journal: Bioorg. Med. Chem. Lett. / Year: 2016
Title: The discovery and the structural basis of an imidazo[4,5-b]pyridine-based p21-activated kinase 4 inhibitor
Authors: Park, J.K. / Kim, S. / Han, Y.J. / Kim, S.H. / Kang, N.S. / Lee, H. / Park, S.Y. |
|---|
| History | | Deposition | Feb 3, 2016 | Deposition site: RCSB / Processing site: PDBJ |
|---|
| Revision 1.0 | Dec 14, 2016 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Nov 8, 2023 | Group: Data collection / Database references / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
| Revision 1.2 | Oct 23, 2024 | Group: Structure summary / Category: pdbx_entry_details / pdbx_modification_feature |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj





 Assembly
Assembly

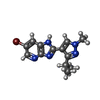
 Mass: 320.188 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H14BrN5
Mass: 320.188 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H14BrN5 Sample preparation
Sample preparation Processing
Processing