 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5fb7: Ligand binding domain 2 of Penicillium marneffei MP1 protein comp... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5fb7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Ligand binding domain 2 of Penicillium marneffei MP1 protein complexed with multiple arachidonic acids | ||||||
 Components Components | Envelope glycoprotein | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / Ligand binding domain 2 / multiple arachidonic acids | ||||||
| Function / homology | Cell wall mannoprotein 1 / Hydrophobic surface binding protein A / fungal-type cell wall / lipid binding / extracellular region / ARACHIDONIC ACID / Cell wall mannoprotein 1 Function and homology information Function and homology information | ||||||
| Biological species |  Talaromyces marneffei PM1 (fungus) Talaromyces marneffei PM1 (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.5 Å | ||||||
 Authors Authors | Lam, W.H. / Zhang, H. / Hao, Q. | ||||||
 Citation Citation | Journal: Cell Chem Biol / Year: 2017 Title: Talaromyces marneffei Mp1p Is a Virulence Factor that Binds and Sequesters a Key Proinflammatory Lipid to Dampen Host Innate Immune Response Authors: Sze, K.H. / Lam, W.H. / Zhang, H. / Ke, Y.H. / Tse, M.K. / Woo, P.C. / Lau, S.K. / Lau, C.C. / Cai, J.P. / Tung, E.T. / Lo, R.K. / Xu, S. / Kao, R.Y. / Hao, Q. / Yuen, K.Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5fb7.cif.gz | 253.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5fb7.ent.gz | 205.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5fb7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fb/5fb7ftp://data.pdbj.org/pub/pdb/validation_reports/fb/5fb7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5csdSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 16599.871 Da / Num. of mol.: 4 / Fragment: UNP residues 188-342 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Talaromyces marneffei PM1 (fungus) / Gene: GQ26_0022220 / Plasmid: His-SUMO / Production host:  #2: Chemical | ChemComp-ACD / 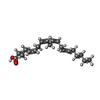  Mass: 304.467 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C20H32O2 Mass: 304.467 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C20H32O2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 427 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 427 / Source method: isolated from a natural source / Formula: H2OSequence details | AUTHORS STATE THAT THE GENEBANK ACCESSION NUMBER KFX52825 IS FOR THIS SEQUENCE. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.08 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 4.6 / Details: PEG 4000, sodium acetate, ammonium acetate |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF  / Beamline: BL17U / Wavelength: 0.9793 Å / Beamline: BL17U / Wavelength: 0.9793 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Mar 26, 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9793 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.5→50 Å / Num. obs: 90586 / % possible obs: 98.9 % / Redundancy: 7.4 % / Rmerge(I) obs: 0.066 / Χ2: 1.088 / Net I/av σ(I): 26.115 / Net I/σ(I): 13 / Num. measured all: 668581 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5CSD Resolution: 1.5→50 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.951 / WRfactor Rfree: 0.2358 / WRfactor Rwork: 0.2039 / FOM work R set: 0.831 / SU B: 3.626 / SU ML: 0.066 / SU R Cruickshank DPI: 0.0839 / SU Rfree: 0.0826 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.084 / ESU R Free: 0.083 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 77.07 Å2 / Biso mean: 30.275 Å2 / Biso min: 16.4 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.5→50 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.539 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
