- PDB-5ezu: Crystal structure of the N-terminal domain of vaccinia virus immu... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 5ezu
Title
Crystal structure of the N-terminal domain of vaccinia virus immunomodulator A46 in complex with myristic acid.
Components
Protein A46
Keywords
VIRAL PROTEIN / immunomodulator / beta sheet / ab initio phasing / vaccinia virus / A46 / fatty acids / myristic acid
Function / homology
Function and homology information
extrinsic component of cytoplasmic side of plasma membrane / protein sequestering activity / symbiont-mediated suppression of host toll-like receptor signaling pathway / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of IRF3 activity / symbiont-mediated suppression of host NF-kappaB cascade Similarity search - Function
Journal: PLoS Pathog / Year: 2016 Title: Vaccinia Virus Immunomodulator A46: A Lipid and Protein-Binding Scaffold for Sequestering Host TIR-Domain Proteins. Authors: Sofiya Fedosyuk / Gustavo Arruda Bezerra / Katharina Radakovics / Terry K Smith / Massimo Sammito / Nina Bobik / Adam Round / Lynn F Ten Eyck / Kristina Djinović-Carugo / Isabel Usón / Tim Skern / Abstract: Vaccinia virus interferes with early events of the activation pathway of the transcriptional factor NF-kB by binding to numerous host TIR-domain containing adaptor proteins. We have previously ...Vaccinia virus interferes with early events of the activation pathway of the transcriptional factor NF-kB by binding to numerous host TIR-domain containing adaptor proteins. We have previously determined the X-ray structure of the A46 C-terminal domain; however, the structure and function of the A46 N-terminal domain and its relationship to the C-terminal domain have remained unclear. Here, we biophysically characterize residues 1-83 of the N-terminal domain of A46 and present the X-ray structure at 1.55 Å. Crystallographic phases were obtained by a recently developed ab initio method entitled ARCIMBOLDO_BORGES that employs tertiary structure libraries extracted from the Protein Data Bank; data analysis revealed an all β-sheet structure. This is the first such structure solved by this method which should be applicable to any protein composed entirely of β-sheets. The A46(1-83) structure itself is a β-sandwich containing a co-purified molecule of myristic acid inside a hydrophobic pocket and represents a previously unknown lipid-binding fold. Mass spectrometry analysis confirmed the presence of long-chain fatty acids in both N-terminal and full-length A46; mutation of the hydrophobic pocket reduced the lipid content. Using a combination of high resolution X-ray structures of the N- and C-terminal domains and SAXS analysis of full-length protein A46(1-240), we present here a structural model of A46 in a tetrameric assembly. Integrating affinity measurements and structural data, we propose how A46 simultaneously interferes with several TIR-domain containing proteins to inhibit NF-κB activation and postulate that A46 employs a bipartite binding arrangement to sequester the host immune adaptors TRAM and MyD88.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Vaccinia virus
Vaccinia virus X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Austria, 1items
Austria, 1items  Citation
Citation





 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj



 Assembly
Assembly


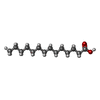
 Mass: 228.371 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28O2
Mass: 228.371 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28O2 Mass: 18.015 Da / Num. of mol.: 57 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 57 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing