 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ynx | ||||||
|---|---|---|---|---|---|---|---|
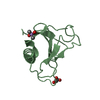
| Title | Structure of YdiE from E. coli | ||||||
 Components Components | Uncharacterized protein YdiE | ||||||
 Keywords Keywords | UNKNOWN FUNCTION / heme uptake / small / homo-dimer / conserved | ||||||
| Function / homology | Hemin uptake protein HemP / Hemin uptake protein hemP / Complement Module, domain 1 / Complement Module; domain 1 / Ribbon / protein homodimerization activity / Mainly Beta / Uncharacterized protein YdiE Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Tame, J.R.H. / Nishimura, K. / Zhang, K.Y.J. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.F / Year: 2015 Title: The crystal and solution structure of YdiE from Escherichia coli Authors: Nishimura, K. / Addy, C. / Shrestha, R. / Voet, A.R. / Zhang, K.Y.J. / Ito, Y. / Tame, J.R.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ynx.cif.gz | 49.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ynx.ent.gz | 34.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4ynx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yn/4ynxftp://data.pdbj.org/pub/pdb/validation_reports/yn/4ynx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2jraS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 7412.448 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: K12 / Gene: ydiE / Plasmid: pET28 / Production host: #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 45 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Description: Crystallization was carried out with full-length YdiE protein, but over 20 residues of each chain are missing from the final electron density map. The Matthews coefficient for the ...Description: Crystallization was carried out with full-length YdiE protein, but over 20 residues of each chain are missing from the final electron density map. The Matthews coefficient for the structure is 1.3, assuming all residues to be present, implying a solvent content of under 10%. If only the ordered residues are present in the crystal, the Matthews coefficient is 2.0, implying a solvent content of 40%. NMR experiments confirm the full-length protein was expressed and purified, but a number of residues are apparently missing from the N-terminus of the crystallized protein. |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 7 / Details: 1.6 M ammonium sulphate, 0.1M Tris |
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL38B1 / Wavelength: 0.9 Å / Beamline: BL38B1 / Wavelength: 0.9 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Mar 11, 2003 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 1.497→30.5 Å / Num. obs: 12710 / % possible obs: 98.4 % / Redundancy: 4.4 % / Rmerge(I) obs: 0.063 / Net I/av σ(I): 28.4 / Net I/σ(I): 19.3 |
| Reflection shell | Resolution: 1.497→1.554 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.346 / Mean I/σ(I) obs: 4.1 / % possible all: 95.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: MOLECULAR DYNAMICS SIMULATION OF PDB 2JRA Resolution: 1.5→30.5 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.915 / SU B: 3.241 / SU ML: 0.054 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.091 / ESU R Free: 0.092 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 75.67 Å2 / Biso mean: 16.45 Å2 / Biso min: 5.37 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.5→30.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.498→1.536 Å / Total num. of bins used: 20
|
