 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1i70 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF RNASE SA Y86F MUTANT | ||||||
 Components Components | GUANYL-SPECIFIC RIBONUCLEASE SA | ||||||
 Keywords Keywords | HYDROLASE / MUTANT | ||||||
| Function / homology |  Function and homology information Function and homology informationribonuclease T1 / ribonuclease T1 activity / hydrolase activity / RNA binding / extracellular region Similarity search - Function | ||||||
| Biological species |  Streptomyces aureofaciens (bacteria) Streptomyces aureofaciens (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / STARTING MODEL WAS USED WITHOUT MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / STARTING MODEL WAS USED WITHOUT MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Sevcik, J. / Urbanikova, L. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2001 Title: Tyrosine hydrogen bonds make a large contribution to protein stability. Authors: Pace, C.N. / Horn, G. / Hebert, E.J. / Bechert, J. / Shaw, K. / Urbanikova, L. / Scholtz, J.M. / Sevcik, J. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE ACCORDING TO THE AUTHOR, THE ORIGINAL SEQUENCE OF THE RNASE SA IN THE SEQUENCE DATABASE ...SEQUENCE ACCORDING TO THE AUTHOR, THE ORIGINAL SEQUENCE OF THE RNASE SA IN THE SEQUENCE DATABASE SWISSPROT ENTRY P05798 IS WRONG. RESIDUE 72 IS THR, NOT CYS. THIS ISSUE IS DISCUSSED IN THE PAPER: SEVCIK J., DAUTER Z., LAMZIN V.S. AND WILSON K.S. (1996), ACTA CRYSTALLOGR. D52, 327-344 ON THE BASIS OF ATOMIC RESOLUTION REFINEMENT. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1i70.cif.gz | 56.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1i70.ent.gz | 41.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1i70.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i7/1i70ftp://data.pdbj.org/pub/pdb/validation_reports/i7/1i70 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1i8vC  1rggS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 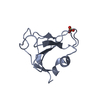
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 10566.492 Da / Num. of mol.: 2 / Mutation: Y86F Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptomyces aureofaciens (bacteria) / Production host: #2: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 305 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 305 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 Details: AMMONIUM SULFATE, MONOSODIUM PHOSPHATE, DISODIUM PHOSPHATE, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||
| Crystal grow | *PLUS Details: used microseeding / PH range low: 7.2 / PH range high: 7 | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LURE  / Beamline: D41A / Wavelength: 1.38 Å / Beamline: D41A / Wavelength: 1.38 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 22, 1999 |
| Radiation | Monochromator: FOCUSING CURVED MONO. / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.38 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→30.7 Å / Num. all: 21828 / Num. obs: 84702 / % possible obs: 97.1 % / Observed criterion σ(F): 0 / Redundancy: 3.9 % / Biso Wilson estimate: 13.9 Å2 / Rmerge(I) obs: 0.061 / Net I/σ(I): 20.8 |
| Reflection shell | Resolution: 1.7→1.76 Å / Rmerge(I) obs: 0.218 / Mean I/σ(I) obs: 7.1 / % possible all: 95.5 |
| Reflection | *PLUS Num. obs: 21859 / Num. measured all: 84702 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: STARTING MODEL WAS USED WITHOUT MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1RGG Resolution: 1.7→30.7 Å / σ(I): 0 / ESU R: 0.13 / ESU R Free: 0.09 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.85 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 30.7 Å / Luzzati sigma a obs: 0.014 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→30.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7→1.76 Å / % reflection obs: 95.5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.7 Å / % reflection Rfree: 5 % / Rfactor Rwork: 0.13 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: p_planar_d / Dev ideal: 0.04 / Dev ideal target: 0.05 |
