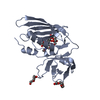
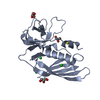
登録情報 データベース : PDB / ID : 4rcoタイトル 1.9 Angstrom Crystal Structure of Superantigen-like Protein, Exotoxin from Staphylococcus aureus, in Complex with Sialyl-LewisX. Putative uncharacterized protein キーワード / / / / / / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / / / / / / / / / / / / / / / / / / 生物種 Staphylococcus aureus subsp. aureus (黄色ブドウ球菌)手法 / / / 解像度 : 1.9 Å データ登録者 Minasov, G. / Nocadello, S. / Shuvalova, L. / Filippova, E. / Halavaty, A. / Dubrovska, I. / Flores, K. / Bagnoli, F. / Falugi, F. / Bottomley, M. ...Minasov, G. / Nocadello, S. / Shuvalova, L. / Filippova, E. / Halavaty, A. / Dubrovska, I. / Flores, K. / Bagnoli, F. / Falugi, F. / Bottomley, M. / Grandi, G. / Anderson, W.F. / Center for Structural Genomics of Infectious Diseases (CSGID) ジャーナル : TO BE PUBLISHED タイトル : 1.9 Angstrom Crystal Structure of Superantigen-like Protein, Exotoxin from Staphylococcus aureus, in Complex with Sialyl-LewisX.著者: Minasov, G. / Nocadello, S. / Shuvalova, L. / Filippova, E. / Halavaty, A. / Dubrovska, I. / Flores, K. / Bagnoli, F. / Falugi, F. / Bottomley, M. / Grandi, G. / Anderson, W.F. / Center for ... 著者 : Minasov, G. / Nocadello, S. / Shuvalova, L. / Filippova, E. / Halavaty, A. / Dubrovska, I. / Flores, K. / Bagnoli, F. / Falugi, F. / Bottomley, M. / Grandi, G. / Anderson, W.F. / Center for Structural Genomics of Infectious Diseases (CSGID) 履歴 登録 2014年9月16日 登録サイト / 処理サイト 改定 1.0 2014年10月1日 Provider / タイプ 改定 1.1 2017年11月22日 Group / カテゴリ / Item 改定 2.0 2020年7月29日 Group Atomic model / Data collection ... Atomic model / Data collection / Database references / Derived calculations / Structure summary カテゴリ atom_site / chem_comp ... atom_site / chem_comp / entity / entity_name_com / pdbx_branch_scheme / pdbx_chem_comp_identifier / pdbx_entity_branch / pdbx_entity_branch_descriptor / pdbx_entity_branch_link / pdbx_entity_branch_list / pdbx_entity_nonpoly / pdbx_molecule_features / pdbx_nonpoly_scheme / pdbx_struct_assembly_gen / struct_asym / struct_conn / struct_ref_seq_dif / struct_site / struct_site_gen Item _atom_site.B_iso_or_equiv / _atom_site.Cartn_x ... _atom_site.B_iso_or_equiv / _atom_site.Cartn_x / _atom_site.Cartn_y / _atom_site.Cartn_z / _atom_site.auth_asym_id / _atom_site.auth_atom_id / _atom_site.auth_comp_id / _atom_site.auth_seq_id / _atom_site.label_asym_id / _atom_site.label_atom_id / _atom_site.label_comp_id / _atom_site.label_entity_id / _atom_site.type_symbol / _chem_comp.name / _chem_comp.type / _pdbx_struct_assembly_gen.asym_id_list / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_comp_id / _struct_ref_seq_dif.details 解説 / Provider / タイプ 改定 2.1 2023年9月20日 Group Data collection / Database references ... Data collection / Database references / Refinement description / Structure summary カテゴリ chem_comp / chem_comp_atom ... chem_comp / chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_DOI / _database_2.pdbx_database_accession
すべて表示 表示を減らす
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Staphylococcus aureus subsp. aureus (黄色ブドウ球菌)
Staphylococcus aureus subsp. aureus (黄色ブドウ球菌) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード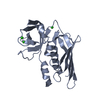








 PDBj
PDBj
 集合体
集合体
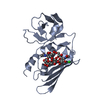



 分子量: 35.453 Da / 分子数: 3 / 由来タイプ: 合成 / 式: Cl
分子量: 35.453 Da / 分子数: 3 / 由来タイプ: 合成 / 式: Cl 分子量: 18.015 Da / 分子数: 465 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 465 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 21-ID-F / 波長: 0.97872 Å
/ ビームライン: 21-ID-F / 波長: 0.97872 Å 解析
解析