 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4r50: Crystal Structure of CNG mimicking NaK-ETPP mutant cocrystallized... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4r50 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of CNG mimicking NaK-ETPP mutant cocrystallized with Li+ | ||||||
 Components Components | Potassium channel protein | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / Alpha helical membrane protein / chimera channel | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.85 Å | ||||||
 Authors Authors | De March, M. / Napolitano, L.M.R. / Onesti, S. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2015 Title: A structural, functional, and computational analysis suggests pore flexibility as the base for the poor selectivity of CNG channels. Authors: Napolitano, L.M. / Bisha, I. / De March, M. / Marchesi, A. / Arcangeletti, M. / Demitri, N. / Mazzolini, M. / Rodriguez, A. / Magistrato, A. / Onesti, S. / Laio, A. / Torre, V. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4r50.cif.gz | 85 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4r50.ent.gz | 65.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4r50.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/r5/4r50ftp://data.pdbj.org/pub/pdb/validation_reports/r5/4r50 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4r6zC  4r7cC  4r8cC  4raiC  4ro2C  3k0dS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||
| Components on special symmetry positions |
| ||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: _ / Ens-ID: 1 / Beg auth comp-ID: LYS / Beg label comp-ID: LYS / End auth comp-ID: ASN / End label comp-ID: ASN / Refine code: _ / Auth seq-ID: 22 - 109 / Label seq-ID: 5 - 92
| ||||||||||||||||||||||||||||||||||||||||||
| Details | Tetrameric channel with filter on the 4-fold axis. |
-Components
| #1: Protein | Mass: 10626.555 Da / Num. of mol.: 2 / Fragment: residues 20-110 / Mutation: D66E, G67T, N68P, F69P Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | ChemComp-MPD / ( |   Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#3: Chemical | ChemComp-GLY / 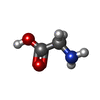  Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H5NO2 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H5NO2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 48 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 48 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | THE AUTHORS STATE THAT THE LITHIUM ION COULD BE AT THE BRIDGE OF TWO WATER MOLECULES IN THE CENTRAL ...THE AUTHORS STATE THAT THE LITHIUM ION COULD BE AT THE BRIDGE OF TWO WATER MOLECULES IN THE CENTRAL CAVITY OF THE K-CHANNELS. SINCE IT HAS ONLY 3 ELECTRONS THE RESOLUTION | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 5 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.38 Å3/Da / Density % sol: 48.26 % |
|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop Details: 40-70% MPD, 20-100mM Glycine, pH 6.5 - 7.5, VAPOR DIFFUSION, HANGING DROP PH range: 6.5 - 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ELETTRA  / Beamline: 5.2R / Wavelength: 1 Å / Beamline: 5.2R / Wavelength: 1 Å | |||||||||||||||
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: Jul 8, 2014 | |||||||||||||||
| Radiation | Monochromator: Si 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | |||||||||||||||
| Reflection twin | Operator: -H,K,-L / Fraction: 0.4761 | |||||||||||||||
| Reflection | Resolution: 2.85→33.81 Å / Num. obs: 4692 / % possible obs: 83 % / Observed criterion σ(F): 1.9 / Observed criterion σ(I): 1.9 | |||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3K0D: chain A without filter and solvent Resolution: 2.85→33.81 Å / Cor.coef. Fo:Fc: 0.966 / Cor.coef. Fo:Fc free: 0.94 / SU B: 17.387 / SU ML: 0.179 / Cross valid method: THROUGHOUT / ESU R Free: 0.319 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.082 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.85→33.81 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|