 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4r7c: Crystal Structure of CNG mimicking NaK-ETPP mutant cocrystallized... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4r7c | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of CNG mimicking NaK-ETPP mutant cocrystallized with DiMethylammonium | ||||||
 Components Components | Potassium channel protein | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / ALPHA-HELICAL membrane protein / NaK-chimera channel in complex with DiMA+ | ||||||
| Function / homology |  Function and homology information Function and homology informationpotassium channel activity / membrane / metal ion binding / identical protein binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | De March, M. / Napolitano, L.M.R. / Onesti, S. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2015 Title: A structural, functional, and computational analysis suggests pore flexibility as the base for the poor selectivity of CNG channels. Authors: Napolitano, L.M. / Bisha, I. / De March, M. / Marchesi, A. / Arcangeletti, M. / Demitri, N. / Mazzolini, M. / Rodriguez, A. / Magistrato, A. / Onesti, S. / Laio, A. / Torre, V. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4r7c.cif.gz | 89.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4r7c.ent.gz | 68.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4r7c.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/r7/4r7cftp://data.pdbj.org/pub/pdb/validation_reports/r7/4r7c | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4r50C  4r6zC  4r8cC  4raiC  4ro2C  3k0dS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||||||||||||||||||||
| Details | The biological tetramer is assembled by a crystallographic 2-fold axis which relates two dimers together. The pore of the tetrameric channel is coincident with the 2-fold crystallographic axis. |
-Components
| #1: Protein | Mass: 10626.555 Da / Num. of mol.: 4 / Fragment: residues 20-110 / Mutation: D66E, G67T, N68P, F69P Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | ChemComp-GLY / 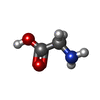  Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 29 / Source method: obtained synthetically / Formula: C2H5NO2 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 29 / Source method: obtained synthetically / Formula: C2H5NO2#3: Chemical | ChemComp-MPD / (   Mass: 118.174 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#4: Chemical | ChemComp-DMN / 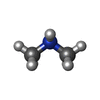  Mass: 45.084 Da / Num. of mol.: 25 / Source method: obtained synthetically / Formula: C2H7N Mass: 45.084 Da / Num. of mol.: 25 / Source method: obtained synthetically / Formula: C2H7N#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 6 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.42 Å3/Da / Density % sol: 49.16 % |
|---|---|
| Crystal grow | pH: 6.5 Details: CNG-ETPP(DiMA+) cocrystals grown in 40-44% MPD, 100mM MES pH6.5 and 20-25 mM Glycine, VAPOR DIFFUSION, HANGING DROP |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ELETTRA  / Beamline: 5.2R / Wavelength: 1 / Beamline: 5.2R / Wavelength: 1 |
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: May 6, 2014 |
| Radiation | Monochromator: SI 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→47.84 Å / Num. obs: 18763 / % possible obs: 99.6 % / Observed criterion σ(I): 2.1 |
| Reflection shell | Resolution: 2.3→47.84 Å / Rmerge(I) obs: 0.071 / Mean I/σ(I) obs: 4.3 / % possible all: 99.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3K0D Resolution: 2.3→47.84 Å / σ(F): 2.1 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→47.84 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|