 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4lrt: Crystal and solution structures of the bifunctional enzyme (Aldol... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4lrt | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal and solution structures of the bifunctional enzyme (Aldolase/Aldehyde dehydrogenase) from Thermomonospora curvata, reveal a cofactor-binding domain motion during NAD+ and CoA accommodation whithin the shared cofactor-binding site | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | Lyase/OXIDOREDUCTASE / Rosmmann Fold / TIM barrel / Dehydrogenase / aldolase / Lyase-OXIDOREDUCTASE complex | |||||||||
| Function / homology |  Function and homology information Function and homology information4-hydroxy-2-oxovalerate aldolase / 4-hydroxy-2-oxovalerate aldolase activity / 2-isopropylmalate synthase activity / acetaldehyde dehydrogenase (acetylating) / acetaldehyde dehydrogenase (acetylating) activity / L-leucine biosynthetic process / NAD binding / manganese ion binding Similarity search - Function | |||||||||
| Biological species |  Thermomonospora curvata (bacteria) Thermomonospora curvata (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | |||||||||
 Authors Authors | Fischer, B. / Branlant, G. / Talfournier, F. / Gruez, A. | |||||||||
 Citation Citation | Journal: To be Published Title: Crystal and solution structures of the bifunctional enzyme (Aldolase/Aldehyde dehydrogenase) from Thermomonospora curvata, reveal a cofactor-binding domain motion during NAD+ and CoA ...Title: Crystal and solution structures of the bifunctional enzyme (Aldolase/Aldehyde dehydrogenase) from Thermomonospora curvata, reveal a cofactor-binding domain motion during NAD+ and CoA accommodation whithin the shared cofactor-binding site Authors: Fischer, B. / Branlant, G. / Talfournier, F. / Gruez, A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4lrt.cif.gz | 544 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4lrt.ent.gz | 450.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4lrt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lr/4lrtftp://data.pdbj.org/pub/pdb/validation_reports/lr/4lrt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4lrsSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj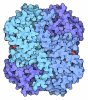






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 4 molecules ACBD
| #1: Protein | Mass: 37326.852 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermomonospora curvata (bacteria) / Strain: DSM 43183 / Gene: Tcur_0536 / Plasmid: pET28b / Production host: References: UniProt: D1A3K8, 4-hydroxy-2-oxovalerate aldolase #2: Protein | Mass: 34127.828 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermomonospora curvata (bacteria) / Strain: DSM 43183 / Gene: Tcur_0535 / Plasmid: pET28b / Production host: References: UniProt: D1A3K7, acetaldehyde dehydrogenase (acetylating) |
|---|
-Non-polymers , 7 types, 1379 molecules 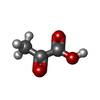




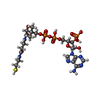

| #3: Chemical |  Mass: 88.062 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H4O3 / References: 4-hydroxy-2-oxovalerate aldolase Mass: 88.062 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H4O3 / References: 4-hydroxy-2-oxovalerate aldolase#4: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na / References: acetaldehyde dehydrogenase (acetylating) Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na / References: acetaldehyde dehydrogenase (acetylating)#5: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#6: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#7: Chemical |  Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#8: Chemical |  Mass: 767.534 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H36N7O16P3S Mass: 767.534 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H36N7O16P3S#9: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1364 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.54 Å3/Da / Density % sol: 51.52 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop Details: 26-34% PEG 4000 or 3350, 0.1 M Tris, 0.2 M Li2SO4, 0.005 M CoA, VAPOR DIFFUSION, HANGING DROP, temperature 298K PH range: 7.5-9.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 1 / Wavelength: 0.8856 Å / Beamline: PROXIMA 1 / Wavelength: 0.8856 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Apr 11, 2011 |
| Radiation | Monochromator: Kirkpatrick-Baez pair of bi-morph mirrors plus channel cut cryogenically cooled monochromator crystal Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8856 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→46.45 Å / Num. all: 208563 / Num. obs: 812171 / % possible obs: 90 % / Observed criterion σ(F): 3 / Observed criterion σ(I): 3 / Redundancy: 3.89 % / Rmerge(I) obs: 0.033 / Net I/σ(I): 22.53 |
| Reflection shell | Resolution: 1.5→1.54 Å / Redundancy: 3.15 % / Rmerge(I) obs: 0.428 / Mean I/σ(I) obs: 3.04 / % possible all: 52.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 4LRS Resolution: 1.5→46.45 Å / Cor.coef. Fo:Fc: 0.973 / Cor.coef. Fo:Fc free: 0.968 / SU B: 2.166 / SU ML: 0.038 / Cross valid method: THROUGHOUT / ESU R: 0.066 / ESU R Free: 0.065 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.556 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→46.45 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|