Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 2
-
Sample preparation
Crystal
Density Matthews: 3.12 Å3/Da / Density % sol: 60.58 %
Crystal grow
Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 8.4 Details: deoxy-HbTb crystals have been grown using 8-9.5 mg/ml protein mixed with equal amount of 7-12 % w/v PEG 6000, PBS 100 mM, KCl 0.3 M, pH 8.4. Deoxy HbTb crystals were then soaked for several ...Details: deoxy-HbTb crystals have been grown using 8-9.5 mg/ml protein mixed with equal amount of 7-12 % w/v PEG 6000, PBS 100 mM, KCl 0.3 M, pH 8.4. Deoxy HbTb crystals were then soaked for several hours in a deoxygenated, CO saturated, stabilizing solution containing 30% w/v PEG 6000, 100 mM Trsi-HCl pH 8.4, 200 mM KCl, 5 mM dithionite reducing agent and flushed with gaseous CO before data collection, VAPOR DIFFUSION, HANGING DROP, temperature 298.0K
Method to determine structure: FOURIER SYNTHESIS / Resolution: 2.2→72.165 Å / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber Details: DIFFRACTION DATA WERE COLLEXCTED ON TWO SINGLE CRYSTALS. THE TWO DATA SET WERE INDEXED, PROCESSED AND SCALED WITH HKL2000. THE AGREEMENT BETWEEN THE TWO DATA SETS WAS OF THE ORDER OF THE ...Details: DIFFRACTION DATA WERE COLLEXCTED ON TWO SINGLE CRYSTALS. THE TWO DATA SET WERE INDEXED, PROCESSED AND SCALED WITH HKL2000. THE AGREEMENT BETWEEN THE TWO DATA SETS WAS OF THE ORDER OF THE EXPECTED ERRORS (RMERGE 0.069). FOR EACH DATA SET, THE STRUCTURE WAS REFINED WITH SHELX PROGRAM. SINCE ALL THE BASIC FEATURES, SUCH AS THE COORDINATION AT THE HEME IRONS, SUBSTANTIALLY AGREE IN THE TWO STRUCTURES THE TWO DATA SETS WERE MERGED FOR SUCCESSIVE REFINEMENT STEPS. ALL REFLECTION WERE USED FOR REFINEMENT
Rfactor
Num. reflection
Selection details
Rwork
0.219
-
-
obs
0.22
33316
-
Rfree
-
1519
RANDOM
all
-
34835
-
Refinement step
Cycle: LAST / Resolution: 2.2→72.165 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
4484
0
174
37
4695
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Trematomus bernacchii (emerald rockcod)
Trematomus bernacchii (emerald rockcod) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj











 Assembly
Assembly
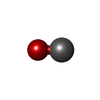
 Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO
Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO
 Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4
Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing