 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4bnc: Crystal structure of the DNA-binding domain of human ETV1 complex... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4bnc | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the DNA-binding domain of human ETV1 complexed with DNA | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | DNA BINDING PROTEIN / DNA-BINDING PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationperipheral nervous system neuron development / sequence-specific double-stranded DNA binding / DNA-binding transcription activator activity, RNA polymerase II-specific / transcription by RNA polymerase II / DNA-binding transcription factor activity, RNA polymerase II-specific / cell differentiation / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / regulation of transcription by RNA polymerase II / chromatin ...peripheral nervous system neuron development / sequence-specific double-stranded DNA binding / DNA-binding transcription activator activity, RNA polymerase II-specific / transcription by RNA polymerase II / DNA-binding transcription factor activity, RNA polymerase II-specific / cell differentiation / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / regulation of transcription by RNA polymerase II / chromatin / positive regulation of transcription by RNA polymerase II / nucleus Similarity search - Function | |||||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 2.9 Å | |||||||||
 Authors Authors | Allerston, C.K. / Cooper, C.D.O. / Krojer, T. / Chaikuad, A. / Vollmar, M. / Froese, D.S. / Arrowsmith, C.H. / Edwards, A. / Bountra, C. / von Delft, F. / Gileadi, O. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2015 Title: Structures of the Ets Domains of Transcription Factors Etv1, Etv4, Etv5 and Fev: Determinants of DNA Binding and Redox Regulation by Disulfide Bond Formation. Authors: Cooper, C.D.O. / Newman, J.A. / Aitkenhead, H. / Allerston, C.K. / Gileadi, O. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4bnc.cif.gz | 74.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4bnc.ent.gz | 54.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4bnc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bn/4bncftp://data.pdbj.org/pub/pdb/validation_reports/bn/4bnc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2yprC  3zp5C  4avpC  4co8C  4unoC  4uuvC C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 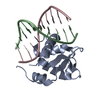
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 12437.209 Da / Num. of mol.: 1 / Fragment: DNA-BINDING DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PNIC28-BSA4 / Production host:  |
|---|---|
| #2: DNA chain | Mass: 3094.042 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) HOMO SAPIENS (human) |
| #3: DNA chain | Mass: 2995.967 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) HOMO SAPIENS (human) |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 7.18 Å3/Da / Density % sol: 82.87 % / Description: NONE |
|---|---|
| Crystal grow | Details: SEMET PROTEIN/DNA WAS CRYSTALLISED IN 20%(W/V) PEG 3350 0.2M POTASSIUM CITRATE. NATIVE PROTEIN/DNA WAS CRYSTALLISED IN 28% LMW PEG SMEAR, 100MM TRIS, PH 8.5, 200MM NACL, 5% GLYCEROL |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.9763 / Beamline: I03 / Wavelength: 0.9763 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: May 13, 2012 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→129.8 Å / Num. obs: 6594 / % possible obs: 99.2 % / Observed criterion σ(I): 2 / Redundancy: 8.9 % / Biso Wilson estimate: 127.18 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 17.6 |
| Reflection shell | Resolution: 2.9→3.06 Å / Redundancy: 9.3 % / Rmerge(I) obs: 1.27 / Mean I/σ(I) obs: 1.9 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIRAS Starting model: NONE Resolution: 2.9→58.02 Å / Cor.coef. Fo:Fc: 0.945 / Cor.coef. Fo:Fc free: 0.9586 / SU R Cruickshank DPI: 0.442 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.508 / SU Rfree Blow DPI: 0.274 / SU Rfree Cruickshank DPI: 0.266
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 123.75 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.899 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→58.02 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→3.24 Å / Total num. of bins used: 5
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
