 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
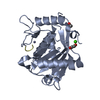
Yorodumi- PDB-4zjf: Crystal structure of GP1 - the receptor binding domain of Lassa virus -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4zjf | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of GP1 - the receptor binding domain of Lassa virus | |||||||||
 Components Components | Glycoprotein | |||||||||
 Keywords Keywords | VIRAL PROTEIN / Arenaviruse / Lassa / receptor binding / glycoprotein | |||||||||
| Function / homology |  Function and homology information Function and homology informationhost cell Golgi membrane / receptor-mediated endocytosis of virus by host cell / host cell endoplasmic reticulum membrane / fusion of virus membrane with host endosome membrane / viral envelope / virion attachment to host cell / host cell plasma membrane / virion membrane / membrane / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Lassa virus Josiah Lassa virus Josiah | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.595 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.595 Å | |||||||||
 Authors Authors | Cohen-Dvashi, H. / Cohen, N. / Israeli, H. / Diskin, R. | |||||||||
 Citation Citation | Journal: J Virol / Year: 2015 Title: Molecular Mechanism for LAMP1 Recognition by Lassa Virus. Authors: Hadas Cohen-Dvashi / Nadav Cohen / Hadar Israeli / Ron Diskin /  Abstract: Lassa virus is a notorious human pathogen that infects many thousands of people each year in West Africa, causing severe viral hemorrhagic fevers and significant mortality. The surface glycoprotein ...Lassa virus is a notorious human pathogen that infects many thousands of people each year in West Africa, causing severe viral hemorrhagic fevers and significant mortality. The surface glycoprotein of Lassa virus mediates receptor recognition through its GP1 subunit. Here we report the crystal structure of GP1 from Lassa virus, which is the first representative GP1 structure for Old World arenaviruses. We identify a unique triad of histidines that forms a binding site for LAMP1, a known lysosomal protein recently discovered to be a critical receptor for internalized Lassa virus at acidic pH. We demonstrate that mutation of this histidine triad, which is highly conserved among Old World arenaviruses, impairs LAMP1 recognition. Our biochemical and structural data further suggest that GP1 from Lassa virus may undergo irreversible conformational changes that could serve as an immunological decoy mechanism. Together with a variable region that we identify on the surface of GP1, those could be two distinct mechanisms that Lassa virus utilizes to avoid antibody-based immune response. IMPORTANCE: Structural data at atomic resolution for viral proteins is key for understanding their function at the molecular level and can facilitate novel avenues for combating viral infections. ...IMPORTANCE: Structural data at atomic resolution for viral proteins is key for understanding their function at the molecular level and can facilitate novel avenues for combating viral infections. Here we used X-ray protein crystallography to decipher the crystal structure of the receptor-binding domain (GP1) from Lassa virus. This is a pathogenic virus that causes significant illness and mortality in West Africa. This structure reveals the overall architecture of GP1 domains from the group of viruses known as the Old World arenaviruses. Using this structural information, we elucidated the mechanisms for pH switch and binding of Lassa virus to LAMP1, a recently identified host receptor that is critical for successful infection. Lastly, our structural analysis suggests two novel immune evasion mechanisms that Lassa virus may utilize to escape antibody-based immune response. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4zjf.cif.gz | 268.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4zjf.ent.gz | 220.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4zjf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zj/4zjfftp://data.pdbj.org/pub/pdb/validation_reports/zj/4zjf | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|







-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 19379.693 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Lassa virus Josiah / Gene: GPC / Production host:  Trichoplusia ni (cabbage looper) / References: UniProt: A0A097F5U5, UniProt: P08669*PLUS Trichoplusia ni (cabbage looper) / References: UniProt: A0A097F5U5, UniProt: P08669*PLUS#2: Polysaccharide | 2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source #3: Sugar | ChemComp-NAG /   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 8 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 8Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.03 Å3/Da / Density % sol: 59.35 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 5.5 Details: 0.1 M BisTris pH 5.0, 0.2 M ammonium acetate pH 7.0, 22 % (w/v) PEG 6000 and 5 % (v/v) PEG 200 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.953 Å / Beamline: ID23-2 / Wavelength: 0.953 Å |
| Detector | Type: DECTRIS PILATUS 2M-F / Detector: PIXEL / Date: Jul 25, 2014 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.953 Å / Relative weight: 1 |
| Reflection | Resolution: 2.595→49.6 Å / Num. obs: 28667 / % possible obs: 99.1 % / Redundancy: 6.8 % / Rmerge(I) obs: 0.102 / Net I/σ(I): 14.3 |
| Reflection shell | Resolution: 2.59→2.68 Å / Rmerge(I) obs: 1.139 / Mean I/σ(I) obs: 1.6 / % possible all: 94.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.595→49.56 Å / SU ML: 0.35 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 27.6 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.595→49.56 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
