regulation of cellular response to hypoxia / negative regulation of receptor signaling pathway via JAK-STAT / RHOBTB3 ATPase cycle / target-directed miRNA degradation / elongin complex / Replication of the SARS-CoV-1 genome / transcription elongation factor activity / VCB complex / Cul5-RING ubiquitin ligase complex / ubiquitin-dependent protein catabolic process via the C-end degron rule pathway ...regulation of cellular response to hypoxia / negative regulation of receptor signaling pathway via JAK-STAT / RHOBTB3 ATPase cycle / target-directed miRNA degradation / elongin complex / Replication of the SARS-CoV-1 genome / transcription elongation factor activity / VCB complex / Cul5-RING ubiquitin ligase complex / ubiquitin-dependent protein catabolic process via the C-end degron rule pathway / Cul2-RING ubiquitin ligase complex / SUMOylation of ubiquitinylation proteins / negative regulation of transcription elongation by RNA polymerase II / Pausing and recovery of Tat-mediated HIV elongation / Tat-mediated HIV elongation arrest and recovery / HIV elongation arrest and recovery / Pausing and recovery of HIV elongation / negative regulation of signal transduction / Tat-mediated elongation of the HIV-1 transcript / Formation of HIV-1 elongation complex containing HIV-1 Tat / Formation of HIV elongation complex in the absence of HIV Tat / ubiquitin-like ligase-substrate adaptor activity / RNA Polymerase II Transcription Elongation / Formation of RNA Pol II elongation complex / negative regulation of TORC1 signaling / RNA Polymerase II Pre-transcription Events / ciliary tip / transcription corepressor binding / negative regulation of autophagy / protein serine/threonine kinase binding / TP53 Regulates Transcription of DNA Repair Genes / positive regulation of cell differentiation / transcription initiation at RNA polymerase II promoter / transcription elongation by RNA polymerase II / Inactivation of CSF3 (G-CSF) signaling / Vif-mediated degradation of APOBEC3G / Evasion by RSV of host interferon responses / Oxygen-dependent proline hydroxylation of Hypoxia-inducible Factor Alpha / cell morphogenesis / Regulation of expression of SLITs and ROBOs / ubiquitin-protein transferase activity / transcription corepressor activity / positive regulation of proteasomal ubiquitin-dependent protein catabolic process / Antigen processing: Ubiquitination & Proteasome degradation / microtubule cytoskeleton / regulation of gene expression / Neddylation / protein-containing complex assembly / Replication of the SARS-CoV-2 genome / cellular response to hypoxia / DNA-binding transcription factor binding / amyloid fibril formation / molecular adaptor activity / ubiquitin-dependent protein catabolic process / proteasome-mediated ubiquitin-dependent protein catabolic process / protein-macromolecule adaptor activity / protein stabilization / cilium / protein ubiquitination / negative regulation of cell population proliferation / negative regulation of gene expression / ubiquitin protein ligase binding / regulation of transcription by RNA polymerase II / regulation of DNA-templated transcription / negative regulation of apoptotic process / positive regulation of DNA-templated transcription / enzyme binding / negative regulation of transcription by RNA polymerase II / endoplasmic reticulum / mitochondrion / proteolysis / nucleoplasm / nucleus / plasma membrane / cytosol Similarity search - Function
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.97903 Å / Relative weight: 1
Reflection
Resolution: 2.8→45 Å / Num. obs: 38647 / % possible obs: 95.5 % / Observed criterion σ(I): 3 / Redundancy: 6.8 % / Biso Wilson estimate: 65.3 Å2 / Rmerge(I) obs: 0.1 / Net I/σ(I): 14.71
Reflection shell
Resolution: 2.8→2.97 Å / Redundancy: 4 % / Rmerge(I) obs: 0.43 / Mean I/σ(I) obs: 2.97 / % possible all: 96.1
-
Processing
Software
Name
Version
Classification
BUSTER
2.10.0
refinement
XDS
datareduction
XDS
datascaling
BUSTER
TNT
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: PVHL54-213-ELOB-ELOC APO STRUCTURE Resolution: 2.8→27.59 Å / Cor.coef. Fo:Fc: 0.935 / Cor.coef. Fo:Fc free: 0.9076 / Cross valid method: THROUGHOUT / σ(F): 0 / SU Rfree Blow DPI: 0.342 Details: IDEAL-DIST CONTACT TERM CONTACT SETUP. ALL ATOMS HAVE CCP4 ATOM TYPE FROM LIBRARY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2358
1960
5.08 %
RANDOM
Rwork
0.1847
-
-
-
obs
0.1873
38613
95.55 %
-
Displacement parameters
Biso mean: 57.68 Å2
Baniso -1
Baniso -2
Baniso -3
1-
3.5479 Å2
0 Å2
0 Å2
2-
-
3.5479 Å2
0 Å2
3-
-
-
-7.0957 Å2
Refinement step
Cycle: LAST / Resolution: 2.8→27.59 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
9894
0
108
193
10195
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
Restraint function
Weight
X-RAY DIFFRACTION
t_bond_d
0.01
10260
HARMONIC
2
X-RAY DIFFRACTION
t_angle_deg
1.23
14016
HARMONIC
2
X-RAY DIFFRACTION
t_dihedral_angle_d
3305
SINUSOIDAL
2
X-RAY DIFFRACTION
t_incorr_chiral_ct
X-RAY DIFFRACTION
t_pseud_angle
X-RAY DIFFRACTION
t_trig_c_planes
199
HARMONIC
2
X-RAY DIFFRACTION
t_gen_planes
1513
HARMONIC
5
X-RAY DIFFRACTION
t_it
10260
HARMONIC
20
X-RAY DIFFRACTION
t_nbd
X-RAY DIFFRACTION
t_omega_torsion
3.1
X-RAY DIFFRACTION
t_other_torsion
21.53
X-RAY DIFFRACTION
t_improper_torsion
X-RAY DIFFRACTION
t_chiral_improper_torsion
1408
SEMIHARMONIC
5
X-RAY DIFFRACTION
t_sum_occupancies
X-RAY DIFFRACTION
t_utility_distance
X-RAY DIFFRACTION
t_utility_angle
X-RAY DIFFRACTION
t_utility_torsion
X-RAY DIFFRACTION
t_ideal_dist_contact
11613
SEMIHARMONIC
4
LS refinement shell
Resolution: 2.8→2.88 Å / Total num. of bins used: 19
Rfactor
Num. reflection
% reflection
Rfree
0.2918
147
5.03 %
Rwork
0.2039
2773
-
all
0.2084
2920
-
obs
-
-
95.55 %
Refinement TLS params.
Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
4.8262
-2.1442
-0.9536
2.0079
1.5456
4.1268
-0.0354
-0.3988
-0.2516
0.1457
-0.0709
-0.079
0.2948
0.0505
0.1063
-0.0236
-0.0461
0.0778
-0.0732
0.0955
-0.2159
-17.9508
2.6397
42.9632
2
3.2217
0.4545
-2.8599
3.1432
0.5551
4.1393
0.0317
-0.0006
-0.2571
-0.1798
-0.175
-0.2414
0.1594
-0.0332
0.1433
0.1092
-0.0176
0.1299
-0.1375
0.0109
-0.1543
-14.5036
-1.4737
25.8336
3
0.8358
0.4805
0.5008
7.1814
-3.6426
3.9341
0.0944
0.1389
0.0165
-0.2869
-0.0362
-0.029
-0.0238
-0.0864
-0.0583
0.0717
0.0017
0.255
-0.1878
-0.0437
-0.1962
-7.5029
19.2555
8.9428
4
1.9465
-2.2289
-1.3523
4.2141
1.8249
5.3809
0.1121
-0.7122
0.0809
0.449
-0.0436
0.482
0.7468
-0.4204
-0.0685
0.0065
-0.1332
0.0594
0.0238
0.0286
-0.2811
28.3426
-1.2324
43.8461
5
2.3592
0.6279
-2.8018
3.486
1.0328
6.9258
-0.0041
-0.019
-0.3311
0.0928
-0.1195
0.0221
0.4415
-0.4236
0.1236
0.0538
-0.0892
0.066
-0.1349
0.0172
-0.176
32.1884
-5.2907
26.0554
6
1.4002
-0.4557
-0.5704
4.8175
-0.9528
3.8262
0.1712
-0.0094
-0.0082
-0.3273
-0.0542
0.0691
-0.3196
-0.1044
-0.1171
0.1146
-0.0178
0.1129
-0.1905
-0.0302
-0.204
39.1557
14.3713
8.9643
7
2.0584
-0.9432
-0.5779
2.3486
0.3506
6.588
-0.1989
-0.3088
0.2375
0.3542
0.1483
0.3342
0.4821
-0.3902
0.0506
0.1488
-0.0117
0.1503
-0.0976
0.0484
-0.3116
32.9284
44.0465
43.404
8
1.227
1.4683
-1.1471
2.855
0.3528
5.0467
0.0026
0.111
0.1986
0.0031
-0.0323
0.3897
0.7178
-0.3611
0.0297
0.2513
-0.0386
0.091
-0.1418
0.0317
-0.2529
34.7201
40.595
25.5755
9
1.3696
-0.6528
0.0746
3.3235
-0.7158
1.465
0.1138
0.003
0.0927
-0.4108
-0.1629
-0.073
0.1874
-0.0611
0.0492
0.317
-0.017
0.1267
-0.1755
0.0111
-0.2916
39.3751
61.6085
8.3231
10
3.3891
-2.291
-0.1933
2.666
1.198
5.761
-0.0909
-0.3306
0.2504
0.2491
-0.0153
-0.0251
0.6023
-0.4515
0.1061
0.0634
-0.1103
0.0926
-0.0998
0.0327
-0.276
-14.4682
47.9837
43.0788
11
1.2212
2.2199
-1.4934
4.7122
0.1178
5.8526
0.0416
0.1526
-0.004
-0.1474
-0.1123
0.1569
0.6648
-0.1999
0.0707
0.1625
-0.0638
0.1258
-0.1353
0.0178
-0.1853
-12.3076
44.9036
25.3383
12
1.4101
0.2451
-0.3371
3.2093
-0.9112
2.7943
0.0742
-0.0086
0.0369
-0.3358
-0.1958
-0.0215
0.058
-0.0024
0.1216
0.1602
0.0146
0.2423
-0.2111
-0.0171
-0.2024
-7.2406
65.5569
8.1626
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection details
1
X-RAY DIFFRACTION
1
CHAINA
2
X-RAY DIFFRACTION
2
CHAINB
3
X-RAY DIFFRACTION
3
CHAINC
4
X-RAY DIFFRACTION
4
CHAIND
5
X-RAY DIFFRACTION
5
CHAINE
6
X-RAY DIFFRACTION
6
CHAINF
7
X-RAY DIFFRACTION
7
CHAING
8
X-RAY DIFFRACTION
8
CHAINH
9
X-RAY DIFFRACTION
9
CHAINI
10
X-RAY DIFFRACTION
10
CHAINJ
11
X-RAY DIFFRACTION
11
CHAINK
12
X-RAY DIFFRACTION
12
CHAINL
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads























 PDBj
PDBj













 Assembly
Assembly







 Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2
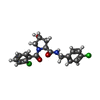
Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 393.264 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C19H18Cl2N2O3
Mass: 393.264 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C19H18Cl2N2O3 Sample preparation
Sample preparation / Beamline: PROXIMA 1 / Wavelength: 0.97903
/ Beamline: PROXIMA 1 / Wavelength: 0.97903  Processing
Processing