 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4a36: Structure of duck RIG-I helicase domain bound to 19-mer dsRNA and... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4a36 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of duck RIG-I helicase domain bound to 19-mer dsRNA and ATP transition state analogue | ||||||
 Components Components |
| ||||||
 Keywords Keywords | RNA BINDING PROTEIN/RNA / RNA BINDING PROTEIN-RNA COMPLEX / SUPERFAMILY 2 RNA HELICASE / ATP AND DSRNA BINDING / ANTIVIRAL SIGNALLING PATHWAY | ||||||
| Function / homology |  Function and homology information Function and homology informationcytoplasmic pattern recognition receptor signaling pathway / antiviral innate immune response / double-stranded RNA binding / RNA helicase activity / single-stranded RNA binding / RNA helicase / hydrolase activity / RNA binding / zinc ion binding / ATP binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  SYNTHETIC CONSTRUCT (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.7 Å | ||||||
 Authors Authors | Kowalinski, E. / Lunardi, T. / McCarthy, A.A. / Cusack, S. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2011 Title: Structural Basis for the Activation of Innate Immune Pattern Recognition Receptor Rig-I by Viral RNA. Authors: Kowalinski, E. / Lunardi, T. / Mccarthy, A.A. / Louber, J. / Brunel, J. / Grigorov, B. / Gerlier, D. / Cusack, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4a36.cif.gz | 265.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4a36.ent.gz | 207.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4a36.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a3/4a36ftp://data.pdbj.org/pub/pdb/validation_reports/a3/4a36 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4a2pSC  4a2qC  4a2vC  4a2wC  4a2xC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




































- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Refine code: 1
NCS ensembles :
NCS oper:
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 63329.492 Da / Num. of mol.: 2 / Fragment: HELICASE DOMAIN, RESIDUES 242-794 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  TRICHOPLUSIA NI (cabbage looper) / References: UniProt: D3TI84 TRICHOPLUSIA NI (cabbage looper) / References: UniProt: D3TI84 |
|---|
-RNA chain , 2 types, 4 molecules RTSU
| #2: RNA chain | Mass: 6031.594 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) SYNTHETIC CONSTRUCT (others) #3: RNA chain | Mass: 6100.713 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) SYNTHETIC CONSTRUCT (others) |
|---|
-Non-polymers , 3 types, 6 molecules 

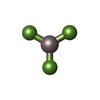
| #4: Chemical |  Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#5: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#6: Chemical |  Mass: 83.977 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: AlF3 Mass: 83.977 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: AlF3 |
|---|
-Details
| Sequence details | EXTRA GAM AT N-TERMINUS AFTER HIS-TAG CLEAVAGE. |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.91 Å3/Da / Density % sol: 58.2 % / Description: NONE |
|---|---|
| Crystal grow | Details: 12 MG/ML, 1.2 TIMES MOLAR RATIO OF 19-MER DSRNA, 4 MM ADP, 2 MM ALF3 AND 0.1 M HEPES PH 7.5, 5% (V/V) ISO-PROPANOL AND 10% (W/V) PEG 4000. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.939 / Beamline: ID14-4 / Wavelength: 0.939 |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: May 5, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.939 Å / Relative weight: 1 |
| Reflection | Resolution: 3.7→50 Å / Num. obs: 18825 / % possible obs: 99.9 % / Observed criterion σ(I): 0 / Redundancy: 7.55 % / Rmerge(I) obs: 0.19 / Net I/σ(I): 10.8 |
| Reflection shell | Resolution: 3.7→3.8 Å / Redundancy: 7.53 % / Rmerge(I) obs: 0.97 / Mean I/σ(I) obs: 2.4 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4A2P Resolution: 3.7→45.95 Å / Cor.coef. Fo:Fc: 0.941 / Cor.coef. Fo:Fc free: 0.893 / SU B: 38.592 / SU ML: 0.567 / Cross valid method: THROUGHOUT / ESU R Free: 0.739 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 97.4 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.7→45.95 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|