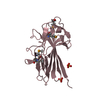
Journal: Open Biol / Year: 2014 Title: The basic keratin 10-binding domain of the virulence-associated pneumococcal serine-rich protein PsrP adopts a novel MSCRAMM fold. Authors: Tim Schulte / Jonas Löfling / Cecilia Mikaelsson / Alexey Kikhney / Karina Hentrich / Aurora Diamante / Christine Ebel / Staffan Normark / Dmitri Svergun / Birgitta Henriques-Normark / Adnane Achour / Abstract: Streptococcus pneumoniae is a major human pathogen, and a leading cause of disease and death worldwide. Pneumococcal invasive disease is triggered by initial asymptomatic colonization of the human ...Streptococcus pneumoniae is a major human pathogen, and a leading cause of disease and death worldwide. Pneumococcal invasive disease is triggered by initial asymptomatic colonization of the human upper respiratory tract. The pneumococcal serine-rich repeat protein (PsrP) is a lung-specific virulence factor whose functional binding region (BR) binds to keratin-10 (KRT10) and promotes pneumococcal biofilm formation through self-oligomerization. We present the crystal structure of the KRT10-binding domain of PsrP (BR187-385) determined to 2.0 Å resolution. BR187-385 adopts a novel variant of the DEv-IgG fold, typical for microbial surface components recognizing adhesive matrix molecules adhesins, despite very low sequence identity. An extended β-sheet on one side of the compressed, two-sided barrel presents a basic groove that possibly binds to the acidic helical rod domain of KRT10. Our study also demonstrates the importance of the other side of the barrel, formed by extensive well-ordered loops and stabilized by short β-strands, for interaction with KRT10.
Mass: 18.015 Da / Num. of mol.: 146 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.53 Å3/Da / Density % sol: 51.41 % / Description: NONE
Crystal grow
Method: vapor diffusion, sitting drop Details: 0.2 M LITHIUM SULFATE, 0.1 M SODIUM ACETATE TRIHYDRATE PH 4.6, 25% PEG4000 (W/V) USING THE SITTING DROP VAPOR-DIFFUSION METHOD FOLLOWED BY MICRO-SEEDING
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.97932 Å / Relative weight: 1
Reflection
Resolution: 2.25→48.35 Å / Num. obs: 61680 / % possible obs: 100 % / Observed criterion σ(I): 3.2 / Redundancy: 4.6 % / Biso Wilson estimate: 36.12 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 14.8
Reflection shell
Resolution: 2.25→2.31 Å / Redundancy: 4.4 % / Rmerge(I) obs: 0.49 / Mean I/σ(I) obs: 3.18 / % possible all: 100
-
Processing
Software
Name
Version
Classification
PHENIX
(PHENIX.REFINE: 1.8.1_1168)
refinement
XDS
datareduction
XSCALE
datascaling
Auto-Rickshaw
phasing
Refinement
Method to determine structure: SAD Starting model: NONE Resolution: 2.25→48.35 Å / SU ML: 0.22 / σ(F): 1.31 / Phase error: 21.32 / Stereochemistry target values: ML Details: NCS AND REFERENCE MODEL TORSION RESTRAINT WERE APPLIED TO CHAINS B/C AND CHAIN A, RESPECTIVELY. CRYSTAL PACKING ANALYSIS REVEALED THAT THE OVERALL MOBILITY OF CHAIN A WAS RELATIVELY HIGHER ...Details: NCS AND REFERENCE MODEL TORSION RESTRAINT WERE APPLIED TO CHAINS B/C AND CHAIN A, RESPECTIVELY. CRYSTAL PACKING ANALYSIS REVEALED THAT THE OVERALL MOBILITY OF CHAIN A WAS RELATIVELY HIGHER COMPARED TO THE MOBILITY OF CHAINS B AND C, AS REFLECTED BY HIGHER OVERALL B-FACTOR VALUES AND LOWER MAP CORRELATION COEFFICIENTS.
Rfactor
Num. reflection
% reflection
Rfree
0.2145
3090
5 %
Rwork
0.186
-
-
obs
0.1874
61669
99.91 %
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL
Refinement step
Cycle: LAST / Resolution: 2.25→48.35 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
3991
0
34
146
4171
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.01
4105
X-RAY DIFFRACTION
f_angle_d
1.47
5573
X-RAY DIFFRACTION
f_dihedral_angle_d
13.9
1446
X-RAY DIFFRACTION
f_chiral_restr
0.077
618
X-RAY DIFFRACTION
f_plane_restr
0.009
698
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
2.25-2.2852
0.282
143
0.2298
2672
X-RAY DIFFRACTION
100
2.2852-2.3226
0.2507
142
0.2207
2656
X-RAY DIFFRACTION
100
2.3226-2.3627
0.2272
138
0.2189
2655
X-RAY DIFFRACTION
100
2.3627-2.4056
0.2505
143
0.2112
2680
X-RAY DIFFRACTION
100
2.4056-2.4519
0.255
137
0.2161
2658
X-RAY DIFFRACTION
100
2.4519-2.502
0.2385
140
0.2157
2660
X-RAY DIFFRACTION
100
2.502-2.5564
0.2616
141
0.2038
2669
X-RAY DIFFRACTION
100
2.5564-2.6158
0.2012
141
0.2063
2658
X-RAY DIFFRACTION
100
2.6158-2.6812
0.2421
138
0.1953
2676
X-RAY DIFFRACTION
100
2.6812-2.7537
0.222
138
0.1979
2654
X-RAY DIFFRACTION
100
2.7537-2.8347
0.2347
143
0.196
2662
X-RAY DIFFRACTION
100
2.8347-2.9262
0.2353
137
0.2014
2671
X-RAY DIFFRACTION
100
2.9262-3.0308
0.2166
137
0.1894
2663
X-RAY DIFFRACTION
100
3.0308-3.1521
0.2521
136
0.1903
2681
X-RAY DIFFRACTION
100
3.1521-3.2956
0.2174
141
0.1931
2648
X-RAY DIFFRACTION
100
3.2956-3.4693
0.2179
140
0.1751
2671
X-RAY DIFFRACTION
100
3.4693-3.6865
0.1806
142
0.1635
2663
X-RAY DIFFRACTION
100
3.6865-3.9711
0.187
143
0.1773
2640
X-RAY DIFFRACTION
100
3.9711-4.3705
0.1956
137
0.1495
2680
X-RAY DIFFRACTION
100
4.3705-5.0023
0.1829
144
0.1371
2668
X-RAY DIFFRACTION
100
5.0023-6.3002
0.179
143
0.1854
2642
X-RAY DIFFRACTION
100
6.3002-48.3614
0.25
146
0.2403
2652
X-RAY DIFFRACTION
99
Refinement TLS params.
Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
3.9521
-0.4428
-1.1893
7.6897
-0.9056
6.2635
-0.1593
-0.4222
-0.5474
0.5145
-0.0253
-0.0609
0.9271
0.4118
0.1604
0.6558
0.0115
0.0601
0.5589
-0.0185
0.5374
44.1787
47.1334
18.8171
2
2.2648
-0.5382
1.6403
1.359
-0.7043
3.7553
0.0497
-0.1495
-0.2381
0.0399
0.1044
0.0317
0.5619
0.1187
-0.114
0.2728
0.061
0.0009
0.1876
0.0083
0.277
18.1786
40.1689
55.9002
3
2.1375
0.5887
-1.8874
1.4096
-1.0419
5.4311
0.1173
0.0232
0.1815
0.0262
0.0768
0.0869
-0.5948
0.2128
-0.1364
0.2136
-0.0397
0.0164
0.1506
-0.0145
0.2347
17.1184
66.306
34.6647
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection details
1
X-RAY DIFFRACTION
1
CHAINA
2
X-RAY DIFFRACTION
2
CHAINB
3
X-RAY DIFFRACTION
3
CHAINC
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 STREPTOCOCCUS PNEUMONIAE (bacteria)
STREPTOCOCCUS PNEUMONIAE (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj Assembly
Assembly



 Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4
 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 18.015 Da / Num. of mol.: 146 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 146 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.97932
/ Beamline: ID29 / Wavelength: 0.97932  Processing
Processing