 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3wif: Crystal structure of anti-prostaglandin E2 Fab fragment 9Cl-PGF2b... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3wif | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of anti-prostaglandin E2 Fab fragment 9Cl-PGF2beta complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | IMMUNE SYSTEM / immunogloblin / Anti-Prostaglandin E2 antibody / Prostaglandin E2 | ||||||
| Function / homology | Immunoglobulins / Immunoglobulin-like / Sandwich / Mainly Beta / Chem-ON5 Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Sugahara, M. / Ago, H. / Saino, H. / Miyano, M. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal structure of anti-Prostaglandin E2 Fab fragment with Prostaglandin E2 Authors: Sugahara, M. / Ago, H. / Saino, H. / Miyano, M. / Kurahashi, Y. / Aoyama, S. / Takehira, M. / Yutani, K. / Yamamoto, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3wif.cif.gz | 116.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3wif.ent.gz | 87.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3wif.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wi/3wifftp://data.pdbj.org/pub/pdb/validation_reports/wi/3wif | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3we6C  3wfhC  3whxC  2ddqS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Antibody | Mass: 23511.332 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Antibody | Mass: 23893.516 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
| #3: Chemical | ChemComp-ON5 / (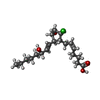  Mass: 372.927 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H33ClO4 Mass: 372.927 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H33ClO4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 744 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 744 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | THE REFERENCE SEQUENCE OF CHAIN A IS BAL50003 AND CHAIN B IS BAL50004 IN GENBANK. |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.53 Å3/Da / Density % sol: 51.43 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: oil microbatch / pH: 5 Details: 0.1M Na acetate, 25%(w/v) PEG3350, pH 5.0, OIL MICROBATCH, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL26B2 / Wavelength: 1 Å / Beamline: BL26B2 / Wavelength: 1 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Oct 25, 2009 |
| Radiation | Monochromator: SI(111) DOUBL CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→35.64 Å / Num. obs: 53235 / % possible obs: 99.2 % / Redundancy: 7 % / Rmerge(I) obs: 0.1 |
| Reflection shell | Resolution: 1.7→1.79 Å / Redundancy: 7 % / Rmerge(I) obs: 0.406 / Mean I/σ(I) obs: 3.8 / Num. unique all: 7287 / % possible all: 94.4 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2DDQ Resolution: 1.7→35.33 Å / Occupancy max: 1 / Occupancy min: 0.35 / FOM work R set: 0.8959 / SU ML: 0.18 / σ(F): 1.4 / Phase error: 17.19 / Stereochemistry target values: ML Details: The structure was refined also with REFMAC5 and CNS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 73.11 Å2 / Biso mean: 20.8515 Å2 / Biso min: 3.63 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→35.33 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 10
|
