 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3sjh: Crystal Structure of a chimera containing the N-terminal domain (... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3sjh | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of a chimera containing the N-terminal domain (residues 8-29) of drosophila Ciboulot and the C-terminal domain (residues 18-44) of bovine Thymosin-beta4, bound to G-actin-ATP-Latrunculin A | ||||||
 Components Components |
| ||||||
 Keywords Keywords | CONTRACTILE PROTEIN / PROTEIN BINDING / protein-protein complex | ||||||
| Function / homology |  Function and homology information Function and homology informationactin monomer sequestering activity / larval central nervous system remodeling / positive regulation of neutrophil migration / negative regulation of actin filament polymerization / negative regulation of neutrophil chemotaxis / cellular response to glucocorticoid stimulus / cytoskeletal motor activator activity / positive regulation of wound healing / myosin heavy chain binding / tropomyosin binding ...actin monomer sequestering activity / larval central nervous system remodeling / positive regulation of neutrophil migration / negative regulation of actin filament polymerization / negative regulation of neutrophil chemotaxis / cellular response to glucocorticoid stimulus / cytoskeletal motor activator activity / positive regulation of wound healing / myosin heavy chain binding / tropomyosin binding / actin filament bundle / troponin I binding / filamentous actin / mesenchyme migration / skeletal muscle myofibril / striated muscle thin filament / actin filament bundle assembly / skeletal muscle thin filament assembly / actin monomer binding / skeletal muscle fiber development / stress fiber / actin filament polymerization / titin binding / regulation of cell migration / actin filament organization / brain development / actin filament / filopodium / protein sequestering activity / negative regulation of inflammatory response / Hydrolases; Acting on acid anhydrides; Acting on acid anhydrides to facilitate cellular and subcellular movement / calcium-dependent protein binding / lamellipodium / cell body / cytoskeleton / protein domain specific binding / hydrolase activity / positive regulation of gene expression / calcium ion binding / magnesium ion binding / : / ATP binding / identical protein binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |   | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.75 Å | ||||||
 Authors Authors | Renault, L. / Husson, C. / Carlier, M.F. / Didry, D. | ||||||
 Citation Citation | Journal: Embo J. / Year: 2012 Title: How a single residue in individual beta-thymosin/WH2 domains controls their functions in actin assembly Authors: Didry, D. / Cantrelle, F.X. / Husson, C. / Roblin, P. / Moorthy, A.M. / Perez, J. / Le Clainche, C. / Hertzog, M. / Guittet, E. / Carlier, M.F. / van Heijenoort, C. / Renault, L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3sjh.cif.gz | 175.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3sjh.ent.gz | 135.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3sjh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sj/3sjhftp://data.pdbj.org/pub/pdb/validation_reports/sj/3sjh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3u8xC  3u9dC  3u9zC  1sqkS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 41862.613 Da / Num. of mol.: 1 / Fragment: UNP residues 3-377 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Protein | Mass: 5988.682 Da / Num. of mol.: 1 Fragment: UNP O97428 residues 8-29, UNP P62326 residues 18-44 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: cib, EG:EG0007.11, CG4944, Dmel_CG4944 / Plasmid: pGEX-6P1 / Production host:  |
-Non-polymers , 4 types, 362 molecules 

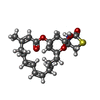

| #3: Chemical | ChemComp-ATP /  Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM |
|---|---|
| #4: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #5: Chemical | ChemComp-LAR /  Mass: 421.550 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H31NO5S Mass: 421.550 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H31NO5S |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 359 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 46.08 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: hanging drop / pH: 6.5 Details: 22% PEG3350, 0.2M MgAcetate pH6.5, 0.45M Guanidine HCl, 1% Dioxane, hanging drop, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.98 Å / Beamline: ID23-2 / Wavelength: 0.98 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Jul 21, 2006 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Highest resolution: 1.75 Å / Num. obs: 44860 / % possible obs: 99.7 % / Observed criterion σ(I): -3 / Biso Wilson estimate: 24.372 Å2 / Rmerge(I) obs: 0.052 / Net I/σ(I): 26.62 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1SQK Resolution: 1.75→26.88 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.947 / WRfactor Rfree: 0.2092 / WRfactor Rwork: 0.1777 / Occupancy max: 1 / Occupancy min: 0.4 / FOM work R set: 0.8859 / SU B: 4.268 / SU ML: 0.063 / SU R Cruickshank DPI: 0.1055 / SU Rfree: 0.1023 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.102 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : RESIDUAL ONLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 54.65 Å2 / Biso mean: 18.827 Å2 / Biso min: 2 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→26.88 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.751→1.796 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
