 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3n56: Crystal Structure of human Insulin-degrading enzyme (IDE) in comp... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3n56 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of human Insulin-degrading enzyme (IDE) in complex with human B-type natriuretic peptide (BNP) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HORMONE / INSULYSIN / INSULINASE / A-BETA DEGRADING ENZYME / CRYPTIDASE / HYDROLASE / HORMONE / DISEASE MUTATION / DIABETES MELLITUS / INSULIN / CARDIAC / SECRETED / PROTEASE / DISULFIDE BOND / METALLOPROTEASE / HUMAN INSULIN-DEGRADNG ENZYME / METAL-BINDING / NATRIURETIC PEPTIDE / NATRIURETIC FACTOR / CARDIOVASCULAR REGULATION / HYDROLASE-HORMONE complex | ||||||
| Function / homology |  Function and homology information Function and homology informationdiuretic hormone activity / body fluid secretion / receptor guanylyl cyclase signaling pathway / insulysin / beta-endorphin binding / cGMP biosynthetic process / ubiquitin recycling / insulin catabolic process / insulin metabolic process / amyloid-beta clearance by cellular catabolic process ...diuretic hormone activity / body fluid secretion / receptor guanylyl cyclase signaling pathway / insulysin / beta-endorphin binding / cGMP biosynthetic process / ubiquitin recycling / insulin catabolic process / insulin metabolic process / amyloid-beta clearance by cellular catabolic process / cardiac conduction system development / positive regulation of renal sodium excretion / hormone catabolic process / bradykinin catabolic process / hormone receptor binding / regulation of vascular permeability / cytosolic proteasome complex / negative regulation of systemic arterial blood pressure / positive regulation of protein binding / insulin binding / positive regulation of urine volume / regulation of aerobic respiration / peptide catabolic process / peroxisomal matrix / amyloid-beta clearance / amyloid-beta metabolic process / Insulin receptor recycling / negative regulation of proteolysis / peptide binding / negative regulation of angiogenesis / : / blood vessel diameter maintenance / protein catabolic process / Peroxisomal protein import / neuropeptide signaling pathway / hormone activity / antigen processing and presentation of endogenous peptide antigen via MHC class I / metalloendopeptidase activity / negative regulation of cell growth / regulation of blood pressure / vasodilation / positive regulation of protein catabolic process / insulin receptor signaling pathway / peroxisome / amyloid-beta binding / virus receptor activity / protein folding / endopeptidase activity / basolateral plasma membrane / cell surface receptor signaling pathway / Ub-specific processing proteases / signaling receptor binding / external side of plasma membrane / protein-containing complex binding / cell surface / protein homodimerization activity / ATP hydrolysis activity / protein-containing complex / mitochondrion / proteolysis / : / extracellular exosome / extracellular region / zinc ion binding / ATP binding / identical protein binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 3.102 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 3.102 Å | ||||||
 Authors Authors | Funke, T. / Guo, Q. / Tang, W.-J. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of human Insulin-degrading enzyme (IDE) in complex with human B-type natriuretic peptide (BNP) Authors: Ralat, L.A. / Funke, T. / Ren, M. / Guo, Q. / Dickey, D.M. / Potter, L.R. / Tang, W.-J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3n56.cif.gz | 396.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3n56.ent.gz | 313.6 KB | Display | PDB format |
| PDBx/mmJSON format | 3n56.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n5/3n56ftp://data.pdbj.org/pub/pdb/validation_reports/n5/3n56 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3cwwS S: Starting model for refinement |
|---|---|
| Similar structure data |


























-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 114560.578 Da / Num. of mol.: 2 / Fragment: UNP residues 42-1019 Mutation: C110L, E111Q, C171S, C178S, C257V, C414L, C573N, C590S, C789S, C812A, C819A, C904S, C966N, C974A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: HCG_1810909, IDE, RP11-366I13.1-001 / Plasmid: PPROEX-H6 / Production host:  #2: Protein/peptide | Mass: 3474.120 Da / Num. of mol.: 2 / Fragment: UNP residues 103-134 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: NPPB / References: UniProt: P16860#3: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#4: Chemical | 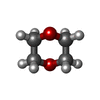  Mass: 88.105 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 88.105 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H8O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 54 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 54 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.86 Å3/Da / Density % sol: 68.17 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 13% PEGMME-5000, 10% TACSIMATE, 10% DIOXANE, 100 mM Na-HEPES, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.9794 Å / Beamline: 19-ID / Wavelength: 0.9794 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Aug 14, 2009 / Details: MIRRORS |
| Radiation | Monochromator: Si(111) AND Si(220)-SAGITALLY FOCUSED DOUBLE CRYSTAL Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9794 Å / Relative weight: 1 |
| Reflection | Resolution: 3.102→50 Å / Num. all: 63372 / Num. obs: 62152 / % possible obs: 96.54 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 2 / Redundancy: 2.5 % / Rmerge(I) obs: 0.20299 / Rsym value: 0.078 / Net I/σ(I): 10.3 |
| Reflection shell | Resolution: 3.102→3.182 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.295 / Mean I/σ(I) obs: 1.8 / Num. unique all: 4642 / Rsym value: 0.462 / % possible all: 98.64 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 3cww Resolution: 3.102→50 Å / Cor.coef. Fo:Fc: 0.926 / Cor.coef. Fo:Fc free: 0.897 / Occupancy max: 1 / Occupancy min: 0.5 / SU B: 16.517 / SU ML: 0.291 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R Free: 0.385 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES: REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 94.43 Å2 / Biso mean: 54.03 Å2 / Biso min: 36.51 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.102→50 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.102→3.182 Å / Total num. of bins used: 20
|
