 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3l4k | ||||||
|---|---|---|---|---|---|---|---|
| Title | Topoisomerase II-DNA cleavage complex, metal-bound | ||||||
 Components Components |
| ||||||
 Keywords Keywords | Isomerase/DNA / TOPOISOMERASE / PROTEIN-DNA COMPLEX / COVALENTLY LINKED COMPLEX / DNA SUPERCOILING / DNA REPLICATION / ATP-binding / DNA-binding / Isomerase / Nucleotide-binding / Nucleus / Phosphoprotein / Isomerase-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationreplication fork progression beyond termination site / DNA replication termination region / regulation of mitotic recombination / sister chromatid segregation / resolution of meiotic recombination intermediates / CENP-A containing chromatin assembly / synaptonemal complex / SUMOylation of DNA replication proteins / DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) activity / telomere maintenance via recombination ...replication fork progression beyond termination site / DNA replication termination region / regulation of mitotic recombination / sister chromatid segregation / resolution of meiotic recombination intermediates / CENP-A containing chromatin assembly / synaptonemal complex / SUMOylation of DNA replication proteins / DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) activity / telomere maintenance via recombination / reciprocal meiotic recombination / DNA topoisomerase (ATP-hydrolysing) / DNA strand elongation involved in DNA replication / DNA topological change / rRNA transcription / chromatin organization / mitochondrion / DNA binding / ATP binding / metal ion binding / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.98 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.98 Å | ||||||
 Authors Authors | Schmidt, B.H. / Burgin, A.B. / Deweese, J.E. / Osheroff, N. / Berger, J.M. | ||||||
 Citation Citation | Journal: Nature / Year: 2010 Title: A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Authors: Schmidt, B.H. / Burgin, A.B. / Deweese, J.E. / Osheroff, N. / Berger, J.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3l4k.cif.gz | 195.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3l4k.ent.gz | 147.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3l4k.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l4/3l4kftp://data.pdbj.org/pub/pdb/validation_reports/l4/3l4k | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3l4jC  2rgrS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 88025.180 Da / Num. of mol.: 1 / Fragment: RESIDUES 421-1177 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: N2244, TOP2, TOR3, YNL088W / Plasmid: PGAL / Production host: |
|---|
-DNA chain , 4 types, 4 molecules BCDE
| #2: DNA chain | Mass: 3269.149 Da / Num. of mol.: 1 / Source method: obtained synthetically |
|---|---|
| #3: DNA chain | Mass: 4675.035 Da / Num. of mol.: 1 / Source method: obtained synthetically |
| #4: DNA chain | Mass: 3109.053 Da / Num. of mol.: 1 / Source method: obtained synthetically |
| #5: DNA chain | Mass: 4529.949 Da / Num. of mol.: 1 / Source method: obtained synthetically |
-Non-polymers , 3 types, 29 molecules 
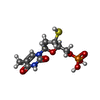

| #6: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn#7: Chemical | ChemComp-TSP / |  Type: DNA linking / Mass: 338.274 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O7PS Type: DNA linking / Mass: 338.274 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O7PS#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 20 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.21 % Description: THE STRUCTURE FACTOR FILE CONTAINS FRIEDEL PAIRS |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 20% 1,4-BUTANEDIOL, 0.1M SODIUM ACETATE, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.3.1 / Wavelength: 1.2823 Å / Beamline: 8.3.1 / Wavelength: 1.2823 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Oct 18, 2009 |
| Radiation | Monochromator: DOUBLE FLAT CRYSTAL, SI (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.2823 Å / Relative weight: 1 |
| Reflection | Resolution: 2.98→50 Å / Num. all: 19750 / Num. obs: 18071 / % possible obs: 91.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.3 % / Biso Wilson estimate: 72.6 Å2 / Rmerge(I) obs: 0.102 / Net I/σ(I): 10.6 |
| Reflection shell | Resolution: 2.98→3.07 Å / Redundancy: 4.4 % / Rmerge(I) obs: 0.538 / Mean I/σ(I) obs: 2.1 / Num. unique all: 1827 / % possible all: 94 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 2RGR Resolution: 2.98→49.023 Å / SU ML: 0.42 / σ(F): 1.9 / Stereochemistry target values: ML / Details: THE FRIEDEL PAIRS WERE USED FOR PHASING
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 30.228 Å2 / ksol: 0.266 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 89.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.98→49.023 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION
|
