 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3hea | ||||||
|---|---|---|---|---|---|---|---|
| Title | The L29P/L124I mutation of Pseudomonas fluorescens esterase | ||||||
 Components Components | Arylesterase | ||||||
 Keywords Keywords | HYDROLASE / alpha/beta hydrolase / esterase / covalent adduct / tetrahedral intermediate / Oxidoreductase / Peroxidase | ||||||
| Function / homology |  Function and homology information Function and homology informationarylesterase / Oxidoreductases / arylesterase activity / peroxidase activity Similarity search - Function | ||||||
| Biological species |  Pseudomonas fluorescens (bacteria) Pseudomonas fluorescens (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Kazlauskas, R.J. / Schrag, J.D. / Cheeseman, J.D. / Morley, K.L. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2010 Title: Switching catalysis from hydrolysis to perhydrolysis in Pseudomonas fluorescens esterase. Authors: Yin de, L.T. / Bernhardt, P. / Morley, K.L. / Jiang, Y. / Cheeseman, J.D. / Purpero, V. / Schrag, J.D. / Kazlauskas, R.J. #1: Journal: Angew.Chem.Int.Ed.Engl. / Year: 2005 Title: Molecular basis of perhydrolase activity in serine hydrolases. Authors: Bernhardt, P. / Hult, K. / Kazlauskas, R.J. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2004Title: Structure of an aryl esterase from Pseudomonas fluorescens. Authors: Cheeseman, J.D. / Tocilj, A. / Park, S. / Schrag, J.D. / Kazlauskas, R.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3hea.cif.gz | 349.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3hea.ent.gz | 285.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3hea.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/he/3heaftp://data.pdbj.org/pub/pdb/validation_reports/he/3hea | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3hi4C  1va4S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
|
-Components
| #1: Protein | Mass: 29977.912 Da / Num. of mol.: 6 / Mutation: L29P/L124I Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas fluorescens (bacteria) / Production host: #2: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-EEE / 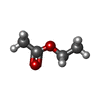  Mass: 88.105 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 88.105 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C4H8O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1057 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1057 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Nonpolymer details | THE C1 CARBON OF EEE BECOMES SP3 RATHER THAN SP2 UPON REACTION WITH SER94. O1 BECOMES SINGLE BONDED ...THE C1 CARBON OF EEE BECOMES SP3 RATHER THAN SP2 UPON REACTION WITH SER94. O1 BECOMES SINGLE BONDED AND ACQUIRES A NEGATIVE CHARGE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.7 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 1.7 M ammonium phosphate, 0.1 M Na,K phosphate, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X29A / Wavelength: 1.1 Å / Beamline: X29A / Wavelength: 1.1 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Mar 27, 2005 / Details: double crystal monochrometer |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→48.1 Å / Num. all: 231042 / Num. obs: 231042 / % possible obs: 96.5 % / Redundancy: 2.62 % / Biso Wilson estimate: 23.2 Å2 / Rsym value: 0.056 / Net I/σ(I): 10.5 |
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 2.54 % / Mean I/σ(I) obs: 3 / Num. unique all: 22477 / Rsym value: 0.313 / % possible all: 93.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1VA4 Resolution: 1.9→48.1 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.947 / SU B: 2.664 / SU ML: 0.077 / Cross valid method: THROUGHOUT / ESU R: 0.11 / ESU R Free: 0.106 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.529 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→48.1 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Ens-ID: 1 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.949 Å / Total num. of bins used: 20
|
